1. Introduction
Peptic ulcer disease, which includes both gastric and duodenal ulcers, markedly affects the global population and has been associated with high morbidity and mortality over the past century [
1,
2]. In gastric ulcers, the balance between protective factors—such as mucus, bicarbonate, prostaglandins (PG), and antioxidant enzymes—and aggravating factors—such as gastric acid, pepsin, Helicobacter pylori infection (
H. pylori), nonsteroid anti-inflammatory drugs (
NSAIDs), smoking, alcohol, and oxidative stress—is disrupted. Ulcerations and mucosal injuries result from this disruption [
1,
3]. Excessive consumption of alcohol negatively affects the gastrointestinal system. Alcohol-induced stomach ulcer formation is hypothesized to be caused by reduced PG synthesis, increased cyclooxygenase (COX), lipoxygenase (LOX), cytokines, and reactive oxygen species (ROS) [
4,
5]. High alcohol consumption generally weakens gastric mucosal defenses, leading to gastric mucosal injuries and resulting in conditions such as gastritis, gastric ulcers, and gastric cancer [
6].
The pharmacological treatment of ulcers involves the use of antacids, proton pump inhibitors (PPIs), antibiotics, and H2 receptor blockers [
7]. Although some pharmacological treatments (such as PPIs and H2 receptor blockers) for stomach ulcers do not completely heal the ulcers, their long-term and continued use has been reported to increase the risk of hypersensitivity, arrhythmia, hypomagnesemia, and gastric cancer [
7,
8,
9]. Therefore, the search for new, safe, and effective strategies for ulcer prevention and treatment has gained importance [
7]. Regulating nutrition and utilizing dietary supplements can also lead to symptom reduction. Consuming essential fatty acids through diet can decrease peptic ulcer incidence, and fresh rice oil reduces gastric ulceration in experimental animal models. When adjusting diets for cases of peptic ulcers, dietary supplements play a crucial role. Powerful antioxidants, such as alpha-lipoic acid and resveratrol, provide protective and therapeutic qualities against experimental ulcers [
10,
11]. Avoiding fatty foods that increase digestive juice is crucial for effective ulcer treatment. Nevertheless, healthy fats are necessary for cell repair and overall health, particularly for the immune system [
12].
Krill oil (KO), extracted from small crustaceans (
Euphausia superba), has attracted attention due to its fatty acid content and beneficial effects on health. It contains omega-3 (
n-3) polyunsaturated fatty acids (PUFA), phospholipids, flavonoids, and astaxanthin (ASX), as well as various vitamins and minerals. Owing to its nutritional composition, KO possesses antioxidant and anti-inflammatory effects [
13,
14], which have resulted from its antioxidant elements, such as vitamin E, choline, and ASX [
15]. KO supplementation reduced inflammatory levels of tumor necrosis factor-alpha (TNF-α) and interleukin-8 (IL-8) in in vitro investigations [
16,
17]. Like KO, fish oil (FO) is a rich dietary source of
n-3 fatty acids. Omega-3 fatty acids, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), are found in seafood, such as fish and algae, and in plant sources, including flaxseed oil, canola oil, walnuts, and purslane. Numerous studies have demonstrated the protective effects of
n-3 fatty acids against cardiovascular disease, cancer, diabetes, and inflammatory illnesses. Leukotrienes, thromboxanes, and PGs made from EPA and DHA have anti-inflammatory properties [
18]. Notably, the bioavailability of EPA and DHA after the intake of KO and FO differs. Gene expression studies using experimental animal models suggested that FO supplementation increased cholesterol synthesis pathway molecules, and KO supplementation regulated more metabolic pathways, such as glucose, fatty acid, and lipid metabolism pathways [
19].
Unlike FO, KO contains ASX, a xanthophyll carotenoid found in various marine animals and some microorganisms. ASX cannot be synthesized in the human body. Experimental studies have suggested that ASX has 100 times more antioxidant activity than alpha-tocopherol and is 10 times stronger than zeaxanthin, lutein, canthaxanthin, and zeaxanthin [
20]. By reducing the synthesis of pro-inflammatory cytokines via the nuclear factor kappa B (NF-κB) pathway, ASX lowers ROS production, hence exhibiting antioxidant effects [
21]. Current evidence in the literature suggests that ASX has therapeutic and preventive benefits for various acute and chronic conditions, including kidney, liver, gastrointestinal, and neurological diseases [
22].
Based on this knowledge, this study was designed to compare the protective effects of nutritional supplementation with KO and FO on tissue integrity, oxidant–antioxidant status, and neutrophil infiltration in inflamed gastric tissue in a rat model of ethanol-induced ulcers. To investigate a possible superiority of ASX-containing KO over ASX-free FO, a group of ASX-only treatments was also added to the study. Similarly, the effects of KO, FO, and ASX supplementation on biochemical and histopathological parameters of liver tissue were analyzed and compared.
4. Discussion
The study findings reveal that supplementation with FO, KO, and ASX provides protective, antioxidant, and anti-inflammatory effects at different levels in rats exposed to ethanol-induced ulcers.
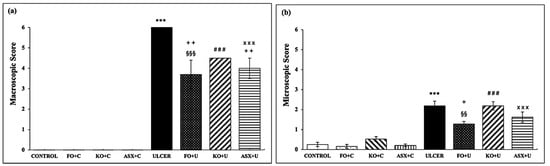
In the present study, the macroscopic data obtained by examining bleeding spots and punctate erosions showed a significant decrease with FO and ASX application, while KO application decreased the lesions but was not statistically significant. Histopathologic examinations showed that FO treatment was more effective in preventing lesion formation than the KO and ASX treatments. Corroborating this study, research using an experimental ethanol-induced ulcer model revealed that ulcerated areas decreased, mucus content increased, and protection against ulceration improved by 67.46% in the group receiving
n-3 supplementation via oral gavage for 14 days [
40]. Bhattacharya et al. examined the effects of FO supplementation at different doses (50, 100, and 200 mg/kg) for 5 days on gastric ulcers and showed that even the lowest dose of FO decreased the ulcer index and gastric acid secretion [
41]. Another study found that FO supplementation for 21 days before aspirin intake did not significantly reduce PGE2 levels or gastric damage in healthy adults [
42]. While our study observed that FO administration had beneficial effects on gastric ulcerations, the effect of FO supplementation was not observed at the level of tissue oxidative stress parameters. In a dextran sulfate sodium-induced ulcerative colitis model, 5% KO supplementation for 4 weeks maintained colon length and significantly improved inflammation-related IL and PG levels in rats, hence suppressing NF-kB activity and cytokine production [
23]. Zhou et al. reported that KO supplementation in mice with dextran sodium sulfate-induced ulcerative colitis for 21 days at a high dose (0.5 g/kg) preserved colonic mucosal integrity, whereas this effect was not observed at a low dose (0.25 g/kg) [
43]. In this study, KO did not show the expected protective effect. Similar to the study by Zhou et al., even though the dose administered in the present study was sufficient, the duration of administration may have been insufficient [
43]. In an ethanol-induced experimental ulcer model, 500 μg/kg ASX supplementation for 21 days protected the stomach mucin layer by 67%, inhibited acid formation by inhibiting the H
+/K
+-ATPase enzyme, and prevented ulcer formation [
44]. ASX supplementation at various doses showed a protective effect against gastric ulcer formation [
28,
45]. In patients diagnosed with
H. pylori, ASX supplementation for 8 weeks did not change inflammatory cytokine levels but enhanced humoral immunity (through upregulation of CD4 expression and downregulation of CD8 expression) [
46]. The different results in these studies show that the optimal doses and application times of FO, KO, and ASX in experimental animal studies remain unclarified.
The stomach and upper gastrointestinal tract are the main sites of ethanol metabolism. Ethanol metabolism generates free radicals, especially superoxide radicals, and promotes lipid peroxidation. Additionally, gastric mucosal injury after ethanol exposure stimulates neutrophils and increases oxygen radicals. Neutrophil infiltration is a major result of gastric injury. Both types of damage can be assessed by measuring tissue-associated MDA levels and MPO activity, respectively [
47]. In this study, ethanol-induced gastric injury significantly increased MDA levels and MPO activity in the ulcer group, and KO or FO supplementation to the ulcer groups failed to significantly reduce lipid peroxidation occurrence. However, FO supplementation significantly reduced neutrophil-related MPO activity in the FO + U group. Shaaban et al. showed that oral omega-3 supplementation at different doses (75 and 150 mg/kg) for 12 weeks markedly reduced serum MDA levels in rats with hepatic fibrosis [
48]. In a TNBS-induced colitis model, rectal administration of FO improved serum and tissue MDA levels but did not result in a significant change [
49]. In another study, FO administration with a high-fat diet for 8 weeks did not change serum MDA levels in hypercholesterolemic rats [
50]. In a gentamicin-mediated nephrotoxicity model, KO supplementation had no effect on tissue MDA and total antioxidant capacity but caused changes at the histopathological level [
51].
KO supplementation decreased serum MDA levels in rats with ischemia-reperfusion injury [
52]. KO can inhibit lipid oxidation [
53,
54] and its supplementation in healthy adults (2 g/day for 6 weeks) did not change plasma TBARS, IL-6, IL-7, and IFNγ levels [
55]. As seen in the literature, the effects of FO and KO on lipid peroxidation vary. Furthermore, FO supplementation can reduce gastric inflammation after ethanol-induced ulcer formation. Experimental studies have shown that FO supplementation reduced inflammatory cytokines, such as TNF-α, IL-1β, IL-6, MIP-1α, MCP-1, and leukotriene B
4 [
56], and attenuated macrophage infiltration, apoptosis, and NO content [
57]. In this study, although KO was expected to be superior to FO and ASX supplements due to its EPA-DHA and ASX content, this effect did not manifest in MDA and GSH levels or MPO activity. As seen in the literature, different results have been obtained. This suggests that the effects of duration of KO application and application dose on tissues occur through different molecular pathways. Ulven et al. concluded that FO upregulated the cholesterol synthesis pathway and this difference in biological effect may be caused by the various structures of phospholipids in KO and triglyceride in FO [
19].
ASX supplementation in various ulcer models yielded increased antioxidant enzyme activities—such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPX)—and decreased lipid peroxidation levels in the ulcer group [
28,
44,
45,
58]. The ASX molecule has a structure containing hydroxyl and keto groups responsible for high antioxidant properties on each ionone ring. Therefore, it inhibits radicals in the cell membrane and scavenges radicals via their terminal rings in the outer and inner parts of the cell membrane. In addition, the oxyfunctional group in carotenoids has high antioxidant activity [
59,
60]. In the present study, ASX inhibited ethanol-induced ulcer oxidative stress due to its strong antioxidant capacity and showed a protective effect against lesions. This is evidenced by the decreased tissue MDA and MPO values and increased GSH levels in the ASX-treated ulcer-induced group. The protective effect of ASX against gastric lesions may be due to its ability to inhibit the H
+/K
+-ATPase enzyme, which markedly contributes to ulcer pathogenesis [
41].
GSH is an antioxidant molecule that forms spontaneously within cells. It plays many biological roles, including DNA and protein synthesis, regulation of enzyme activities, and intracellular and extracellular transport, and it is also closely associated with the antioxidant system [
61]. In the present study, intracellular GSH levels decreased in the ulcer group but did not change significantly in rats receiving KO, FO, and ASX supplementation. In our ethanol-induced ulcer model, the experimental animals were sacrificed 1 h after ethanol administration, which may not be sufficient for glutathione formation [
62,
63]. Actually, the decrease of GSH levels in gastric tissues may be due to its consumption during the oxidative stress induced by alcohol. According to our findings, this result was similar to the study by İpek et al. [
32]. In the same study, it was shown that the application of metformin, an antidiabetic drug, to rats increased SOD enzyme activity, which catalyzes the conversion of superoxide anion to H
2O
2, in gastric samples. In our study, intensive FO, KO, and ASX administration before ulcer formation did not have a stimulating effect on gastric tissue to increase GSH synthesis. However, in various inflammatory settings, FO, KO, and ASX were reported to have antioxidant properties beyond their effects. In an experimentally developed cold restraint stress model, it has been demonstrated that FO application reduces SOD enzyme activity in a dose-dependent manner, while simultaneously increasing the activities of CAT and GPX enzymes [
41]. Among these enzymes, CAT is one of the key enzymes that catalyzes the conversion of H
2O
2 to water, and GPX aims to reduce oxidative stress damage by forming GSH to oxidized GSSG. Similarly, it has been reported that high doses of KO in a different ethanol-induced ulcer model decreased SOD enzyme activity [
64]. A comprehensive experimental ulcer study examining antioxidant enzyme activities has shown that ASX application increases SOD, CAT, and GPX enzyme activities in a dose-dependent manner [
44]. These studies indicate that GSH formation and the GSH-mediated enzyme cycle are inhibited and/or consumed with ulcer formation but can be partially restored with FO, KO, or ASX application. Although our study does not examine GSH-mediated enzyme activities, but the observed trend of increased GSH levels in the gastric tissue of groups treated with FO, KO, and ASX, along with the reductions in lipid peroxidation, can be interpreted as an attempt to restore the GSH-mediated enzymatic mechanism.
In this study, CL analysis revealed that mitochondrial free radicals associated with both inflammation and ischemic damage increased in the ethanol-induced experimental ulcer model, indicating that ROS and reactive nitrogen species (RNS) increased after ethanol-induced ulcer formation. In addition, FO, KO, and ASX supplementation to ulcer-created gastric tissues reduced all species of ROS and RNS. Şehirli et al. studied the effects of α-lipoic acid on ethanol-induced gastric ulcers and found that ethanol administration increased luminol- and lucigenin-mediated CL levels. This finding, corroborating ours, supports the idea that ethanol-induced gastric damage generates toxic oxygen metabolites, such as hydroxyl radicals, H
2O
2, hydroperoxyl, hypochlorite, and superoxide radicals [
47]. Luminol- and lucigenin-mediated ROS formation in the gastric tissues of rats treated with FO and KO were significantly decreased relative to the non-supplemented ulcer group. ASX administration prevented ROS formation caused by ethanol-mediated ulcer damage with its strong antioxidant effect and reduced ROS formation to the basal level. Notably, ethanol rapidly passes through the gastric mucosa, causing endothelial damage in blood vessels through membrane injury and increasing mucosal permeability. Vascular and microvascular changes accelerate the formation of gastric ulcers. ASX pretreatment increases mucus production by blocking the H
+/K
+-ATPase proton pump, thereby protecting the gastric mucosal layer from free radical damage and gastric acid secretion [
65]. In an experimental ethanol/HCl ulcer model where a single dose (30 or 100 mg/kg BW; 1 h) of ASX was applied, it was shown that the loss of epithelial cells in gastric tissue decreased [
58]. In a similar experimental study on a pancreatitis model in rats, Gürler et al. found that a single dose (40 mg/kg BW; 1 h) of ASX administered orogastrically decreased luminol- and lucigenin-enhanced ROS formation and increased GSH content [
66]. In another experimental ulcer model, ASX supplementation had a protective effect against ulcers due to its antioxidant properties and provided 23 times more LOX enzyme inhibition compared to PPI omeprazole [
44], indicating its powerful anti-inflammatory effect. A study exploring various hydrophilic and lipophilic antioxidants implicated ASX as the molecule with the strongest singlet oxygen scavenging activity [
67]. Owing to its chemical structure, ASX is more stable than many other antioxidant molecules, neutralizes ROS in mitochondria, and protects the cell membrane against oxidative stress damage [
68,
69]. ASX localizes not only to the cell membrane but also to the mitochondrial membrane, where it affects cytochrome c and pro-apoptotic mechanisms, preventing the increase of free radicals. Moreover, through an indirect pathway, it activates antioxidant signaling pathways, regulating mitochondrial redox status and maintaining membrane integrity [
65]. In this study, ROS levels were significantly decreased thanks to the strong antioxidant potential of ASX, in support of the literature.
Nitric oxide is synthesized by nitric oxide synthase (NOS) in three different ways. Endothelial NOS (eNOS)-mediated NO causes vasodilation of endothelial cells, and neuronal NOS (nNOS)-mediated NO acts as a second messenger molecule in neurons. Inducible NOS (iNOS)-mediated NO is synthesized from leukocytes to promote inflammation. Mitochondrial superoxide radicals are the most important product of mitochondrial redox state change, especially in ischemia and inflammation. Under severely toxic conditions, the peroxynitrite radical, a more toxic molecule, is formed in the presence of superoxide radicals and NO [
70]. In the present study, both NO and peroxynitrite radicals increased in the gastric tissue of rats with ethanol-induced ulceration. Histopathologic examination of the ulcers revealed cell infiltration, migration of leukocytes to the lesion areas, and increased inflammation. Our findings suggest that NO release is mostly mediated by leukocyte-mediated iNOS. In an acetic acid-induced colitis model, acetic acid administration increased luminol, lucigenin, NO, and peroxynitrite CL levels [
71].
In the present study, the animals were euthanized 1 h after the experimental ulcer model was established, and the effect of ethanol on liver function tests was examined within this 1 h period. As a result of the regular use of KO, FO, and ASX, changes that may occur in triglyceride and fat metabolism can be seen in the blood. Therefore, the supplementation and ulcer groups were evaluated together. KO and FO treatments decreased serum total cholesterol, triglyceride, and LDL levels relative to the non-supplemented groups. However, while the FO administration results were significant, those of KO administration were not. Vigerust et al. found that in C57BL/6hTNF-α transgenic mice, KO was superior to FO in reducing triacylglycerol levels. Although the difference was not statistically significant, total cholesterol, HDL cholesterol, and LDL cholesterol levels were found to be lower in the group receiving the FO-rich diet [
72]. In a double-blind, crossover, placebo-controlled, randomized trial, Ramprasath et al. found that KO supplementation for 4 weeks in 24 healthy adults increased total and LDL cholesterol levels, while serum triglyceride and HDL cholesterol levels did not change with either KO or FO treatment—findings that may have resulted from the participants being normolipidemic individuals [
73]. Some studies have uncovered that KO administration in adults yielded hypolipidemic activity, with this effect being pronounced in participants with hyperlipidemia [
74,
75]. Tillander et al. administered C57BL/6J mice with a diet containing high levels of fat (24% fat), FO (5.8% FO), or KO (5.7% KO) for 6 weeks. Plasma total cholesterol, triacylglycerol, and phospholipids were significantly decreased by FO administration, while KO decreased non-esterified fatty acids. Further analysis revealed that the effects of FO and KO on lipid metabolism were mediated by different pathways. While FO inhibited PPAR-α activation in the liver and intestine, KO administration decreased the expression and activity of fatty acid synthase, acetyl-CoA carboxylase, and HMG-CoA reductase enzymes. PPAR-α activation increases the hepatic expression of lipogenic genes, with a simultaneous increase in fatty acid synthesis and TAG accumulation. In addition this study found a decrease in the gene expression of proteins involved in cholesterol and fatty acid synthesis due to KO administration [
76]. These differences in effect may be attributed to differences in the structure of
n-3 fatty acids found in KO and FO [
73,
76]. Current findings in the literature support the results of this study.
Liver functional enzymes remained unchanged after four-week treatment with FO, KO, and ASX supplementation. Sistilli et al. found that KO supplementation reduced ALT levels and hepatic steatosis in mice fed a diet containing 30% triglyceride form
n-3 (FO) and KO for 24 weeks. However, no significant difference was observed in AST levels [
77]. Hwang et al. found that KO administration together with metformin decreased ALT, AST, and ALP levels and provided blood glucose homeostasis in obese mice fed a high-fat diet for 12 weeks [
78]. These studies were conducted on subjects with hepatic damage or fatty deposits. Notably, in the present study, KO supplementation increased liver AST enzyme activity, which is bound to the mitochondria of hepatocytes. This may be explained by the effects of KO on the expression of mitochondrial enzyme activities [
76].