1. Introduction
Over the last decade, the field of oncolytic virus (OV) therapy has experienced remarkable progress, highlighted by significant clinical advancement and expanding therapeutic applications. The approval of the first oncolytic virus, a recombinant herpes simplex virus (T-VEC), by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of metastatic melanoma, nearly ten years ago, marked a pivotal advancement [
1]. However, the marketing progress since then has been slow, and the next OV therapy approval occurred in 2021, this time for malignant glioma, and sanctioned solely in Japan [
2]. This trajectory, although slower than expected, is nevertheless underscored by the exponential growth in clinical studies and has resulted in a growing accumulation of clinical data that validate the immunotherapeutic potential of oncolytic virus therapies, although adenoviruses have been by far the most commonly used OV vector, and melanoma, glioma, and gastrointestinal cancer are among the most targeted types of cancer [
3]. These studies have generally demonstrated that OVs are well tolerated, even in an immune-suppressed setting, with mostly mild and acceptable toxicities having been reported [
4].
Despite these important advancements, most OV candidates are still in the early stages of preclinical or clinical (phase I or II) development [
5]. Nevertheless, it is becoming increasingly evident that OV approaches hold immense promise as cancer therapies, owing to their potent therapeutic effects and favorable safety profile, primarily attributed to their tumor-selective replication mechanisms. A critical aspect responsible for the tumor selectivity of RNA-based oncolytic viruses is the acquired deficiencies in antiviral type I interferon signaling (IFN) pathways within tumor cells, contributing to evasion from immune surveillance, while also facilitating viral propagation [
6]. Conversely, the intact antiviral defense mechanisms in healthy cells serve to restrain viral replication by inducing an orchestrated transcriptional response mediated by IFN-signaling elements [
7,
8]. In addition their direct oncolytic effects, it has more recently been shown that OVs can additionally modulate the tumor microenvironment, transforming it from an immunologically “cold” to “hot” milieu [
9]. This shift is attributed to innate immune responses against the virus and the phenomenon of immunogenic cell death (ICD), characterized by the release of danger-associated molecular patterns (DAMPs) and tumor-associated antigens (TAAs) upon tumor cell lysis [
10]. The recognition of DAMPs by innate immune cells triggers an intricate cascade of events, culminating in the cross-presentation of TAAs by antigen-presenting cells (APCs) to cytotoxic CD8
+ T cells, thereby eliciting a robust adaptive immune response to target local and distant tumors and micro-metastases [
11]. In contrast to the TCR-dependent activation of T cells, a TCR-independent and cytokine-dependent mechanism of T cell activation as a response to viral infection has been described as bystander activation [
12]. Type I interferon leads to the expansion of CD8
+ T cells; however, the precise mechanism of this activation pathway has not yet been fully elucidated [
13].
While the central role of immune stimulation in oncolytic virotherapy has been described, a comprehensive in vivo characterization of the immune response after OV treatment, encompassing local responses in treated tumors, as well as systemic responses in spleen, draining lymph nodes, blood, and distant tumors is imperative in order to better understand the involvement of different immune cell types and their respective localizations. This would allow a more detailed mechanism of action and characterization of the immune-modulatory aspect of the therapy and the development of rationally designed next-generation vectors and combination therapies to optimize these effects.
We have previously reported on a novel chimeric OV construct, comprising the vesicular stomatitis virus (VSV) backbone and the Newcastle disease virus (NDV) envelope proteins [
14]. This construct demonstrated superior safety and efficacy compared to its parental viruses, offering enhanced anti-tumor immune stimulation through its ability to mediate highly immunogenic cell death via syncytia formation [
14,
15]. Moreover, it was shown to synergize with anti-CTLA-4 treatment and adoptive cell therapy (ACT), resulting in improved tumor responses and prolonged survival [
15,
16].
In this study, we conducted an in-depth characterization of the cellular immune response to oncolytic VSV-NDV using the murine B16OVA system, employing chicken ovalbumin as an immunogenic model antigen in order to distinguish between tumor-associated antigen-specific, virus-specific, and overall T cell responses. The analysis of distant and directly treated tumors revealed a significant increase in activated CD4+ and CD8+ T cells and tumor-specific CD8+ T cells upon intratumoral VSV-NDV treatment, demonstrating the transition from a “cold” to a “hot” tumor microenvironment. In the local TDLNs, we observed an early increase in the influx and activation of all examined immune cell types. Importantly, VSV-NDV treatment also induced a systemic enhancement of T cell responses in peripheral blood and in the spleen, and CD8+ T cell depletion completely reversed VSV-NDV-mediated anti-tumor effects, elucidating a critical mechanism of action of this form of therapy. These results unveil a multifaceted systemic immune response as a crucial component of oncolytic VSV-NDV therapy, thereby supporting this fusogenic virus as a promising platform for cancer immunotherapy.
2. Materials and Methods
2.1. Tumor Cell Lines and Virus
B16OVA and HepG2 cells (generous gifts from Simon Heidegger and Ulrike Protzer, respectively, from Klinikum rechts der Isar, Munich, Germany) were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), minimum essential medium (MEM) non-essential amino acids, and sodium pyruvate. VSV-NDV was generated as described previously [
14]. Virus stocks used for the experiments were produced in adherent AGE1.CR.pIX cells (ProBioGen, Berlin, Germany) and purified by ultracentrifugation over a sucrose gradient. Purified virus was resuspended in PBS after an additional ultracentrifugation step, and stocks were stored at −80 °C until use. Viral titers were determined by TCID50 assay in AGE1.CR.pIX cells.
2.2. Mouse Studies
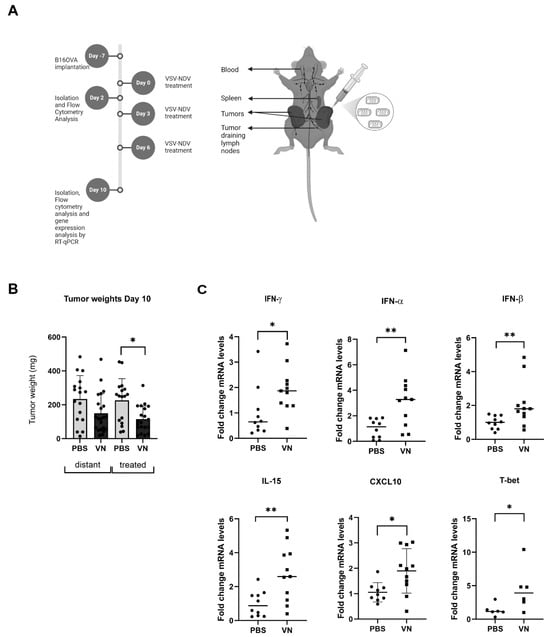
All animal studies were approved by the institute’s commission for preclinical animal research and the regional government commission for animal protection (Regierung von Oberbayern, Munich, Germany). Female C57Bl/6J mice aged 6–8 weeks (Janvier Labs, France) were maintained under specific pathogen-free conditions. A subcutaneous bilateral tumor model was used for survival and mechanistic endpoint analysis. Mice were shaved and then injected subcutaneously with B16OVA into the right (2.4 × 105 cells) and left (1.2 × 105 cells) flanks in a volume of 50 µL of PBS. One week later (day 0), when tumors were visible (tumor volume approximately 20–50 mm3), mice were treated by intratumoral injection of 50 µL of VSV-NDV (1 × 107 TCID50) or PBS into the tumor located on the right lateral flank (injected tumor), repeated on days 3 and day 6. For survival experiments, mice were monitored daily and euthanized when the maximum tumor diameter exceeded 15 mm or tumor rupture occurred, according to the requirements of local regulatory agencies. On days 2 and 10 after the first treatment, additional cohorts of mice were euthanized, tissue samples were collected, and single-cell suspensions were generated. Blood was collected in EDTA-microvettes (Sarstedt), and red blood cell (RBC) lysis was performed with RBC buffer (BioLegend, San Diego, CA, USA). Single-cell suspensions of dispersed tissue were used for flow cytometry. For cell depletion experiments, 100 µg of anti-CD8 antibody or IgG isotype control (both Bio X Cell) were initially given one day pre-treatment start. Complete depletion was confirmed on the first day of virus treatment by flow cytometry, and a survival experiment was conducted as described above.
2.3. RT-qPCR
Tumor tissue was collected and homogenized using a Precellys lysing kit (Bertin Technologies, Montigny-le-Bretonneux, France). RNA was isolated using a Maxwell RSC SimplyRNA tissue kit (Promega, Madison, WI, USA) and the Maxwell RSC instrument (Promega). GoTaq 1-step RT-qPCR Kit (Promega) and a 7500 Real Time PCR system (Applied Biosystems, Foster City, CA, USA) were used to assess transcript abundance. Samples were quantified in duplicates using 50 ng of template RNA and the following cycling conditions: reverse transcription (RT) at 50 °C for 15 min; reverse transcriptase inactivation/hot-start polymerase activation at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s (denaturation), 60 °C (IFN-γ, IL-15, T-bet, IFN-β, and GAPDH) or 53 °C (CXCL10) for 30 s (annealing), and 72 °C for 30 s (extension). GAPDH was used as a housekeeping gene for normalization, and the comparative ΔΔC
T method was used to calculate target gene expression. Primer sequences for amplification of the respective genes are shown in
Table 1.
2.4. In Vitro Co-Culture
HepG2 cells were infected with VSV-NDV at an MOI of 0.1 or 1, in RPMI 1640 Medium GlutaMAX-I (Invitrogen, Waltham, MA, USA), supplemented with 10% heat-inactivated FCS, MEM non-essential amino acids, sodium pyruvate, and β-mercaptoethanol (50 mM) and incubated for 16 h, before adding PBMCs isolated from human blood collected from healthy volunteers (ethics approval under file #318/19 S-SR, granted by the institutional review board of Klinikum rechts der Isar), followed by a 24 h incubation before cells were harvested and stained with flow cytometry antibodies.
2.5. Flow Cytometry
The following antibodies (BioLegend, San Diego, CA, USA) were used for staining for flow cytometry: anti-CD3e (145-2C11), anti-CD4 (GK1.5), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD19c (1D3/CD19), anti-CD44 (IM7), anti-CD69 (H1.2F3), anti-CD86 (GL-1), anti-CD122 (5H4), anti-F4/80 (BM8), anti-H-2Db (KH95), anti-I-A/I-E (M5/114.15.2), anti-IFNγ (XMG1.1), anti-NK-1.1 (PK136), anti-NKG2D(CX5), and anti-PD-1 (29F.1A12). Additionally, the following antibodies (Miltenyi Biotech, Bergisch Gladbach, Germany) were used: anti-CD3 (REA641), anti-CD8 (REA601), anti-CD49a (REA493), anti-CD62L (REA828), and anti-CD69 (H1.2F3), as well as Viobility™ 405/520 Fixable Dye. From Sony, anti-CD49b (DX5) was used and from Invitrogen, anti-CD103 (2E7) was used. The H-2kb OVA Tetramer-SIINFEKL (MBL International, Woburn, MA, USA) was used. We thank the NIH Tetramer Core Facility (contract number 75N93020D00005) for providing I-Ab OVA Tetramer-AAHAEINEA, H-2kb VSV Tetramer-RGYVYQGL, H-2kb OVA Tetramer-SIINFEKL, mouse CD1d Tetramer loaded with the α-GalCer analog PBS57. For human in vitro experiments, the following antibodies (BioLegend) were used: anti-CD3 (UCHT1), anti-CD4 (RPA-T4), anti-CD56 (5.1H11), and anti-CD69 (FN50). Also, anti-CD8 (BW135/80) from miltenyi and anti-IFNγ (B27) from BD Pharmingen were used. For mouse in vitro experiments, anti-IFNγ (XMG1.2) from BioLegend was additionally used.
For experiments including the AAHAEINEA(OVA)-specific tetramer, isolated cells were incubated for 2 h at 37 °C, whereas for experiments including MHC-I tetramers, isolated cells were incubated for 20 min at room temperature with SIINFEKL(OVA)-/(VSV-NP)-specific tetramers. Extracellular staining was performed for 30 min at 4 °C, washed with PBS, optionally fixed and intracellularly stained with BD Cytofix/Cytoperm ™ Fixation/Permeabilization Kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s protocol.
CountBright™ Absolute Counting Beads (Invitrogen™) were used for the calculation of the event count. Flow cytometric measurements were performed using the CytoFLEX S platform (Beckman Coulter Genomics, Brea, CA, USA). The compensation was performed based on staining results from UltraComp beads (Thermo Fisher Scientific, Waltham, MA, USA). All flow cytometry results were analyzed using Flow Jo v10.8 (Ashland, OR, USA) software.
2.6. Statistical Analysis
All data were plotted and analyzed using GraphPad Prism 10.0 (GraphPad, San Diego, CA, USA). Individual data points were compared for statistical significance using an unpaired Student’s t test or ordinary one-way ANOVA for multiple groups, and p values of less than 0.05 were considered to be statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Survival data were plotted as Kaplan–Meier curves, and statistical significance was calculated by log rank test.
4. Discussion
Despite the widespread adoption of immune checkpoint inhibitors (ICI) as the standard of care across various cancer types, such as unresectable or metastatic melanoma, metastatic non-small cell lung cancer (NSCLC), bladder cancer, Hodgkin lymphoma, advanced renal cell carcinoma (RCC), or hepatocellular carcinoma (HCC), a substantial proportion of patients fail to respond due to the prevailing immune-suppressive TME [
17], [
18,
19]. The transition of the immunologically “cold” TME into a “hot” state represents a fundamental objective in current cancer therapy. Strategies aiming at reprogramming the TME include emerging approaches like cytokine therapy, cancer vaccines, and oncolytic viruses [
20]. In the work presented here, we demonstrate that the therapeutic effects of oncolytic VSV-NDV correspond with intricate modulations of immune cell subsets across distinct anatomical compartments. Notably, treatment with VSV-NDV instigated the robust activation and expansion of tumor antigen-specific and effector T cell subsets, which were not confined to the local tumor site, but were also evident systemically, implicating a broader immune activation and highlighting its potential therapeutic utility in metastatic settings.
Notably, when CD8
+ T cells were depleted prior to VSV-NDV treatment in the B16OVA model, the beneficial effects of the virus were abolished, highlighting a central role of this cell type in the mechanism of action of this OV therapy. CXCL10, a chemokine known for its role in immune cell trafficking, and shown to be upregulated in the tumor following VSV-NDV treatment (
Figure 1C), has significant beneficial effects in the TME, particularly in enhancing CD8
+ T cell responses [
21,
22]. We suggest that CXCL10 attracts CD8
+ T cells into the tumor, where they generate robust anti-tumor effects, although the enhanced T cell infiltration in the distant (uninfected) tumor would indicate the presence of an additional, broader immune mechanism. Furthermore, TCR-independent activation of human T cells and NK cells was observed in an in vitro co-culture set up with pre-infected tumor cells, suggesting a multimechanistic immune-stimulatory potential of oncolytic VSV-NDV that could translate to a clinical setting in humans.
These findings are consistent with those observed from other oncolytic viruses. In B16-CD20 melanoma, treatment with oncolytic measles virus led to an increase in CD8
+ T cells in the tumor [
23]. Similarly, treatment with VSV in the B16OVA model led to an increase in CD8+ tumor-infiltrating lymphocytes (TILs) [
24]. VSV-GP, a chimeric variant of VSV, elicited an increase in CD4
+ and CD8
+ T cells within treated B16OVA tumors, albeit generating a weak OVA-specific CD8
+ T cell response [
25]. Previous studies have reported an even weaker antigen-specific immune response for VSV wildtype, attributed to the direct infection and destruction of tumor-associated DCs [
26]. Other oncolytic viruses, such as Zika virus, vaccinia virus, and adenovirus, have similarly been reported to induce cytotoxic T cell infiltration and activation in the tumor microenvironment [
27,
28,
29].
A central hypothesis of our research is that fusogenic oncolytic virus vectors mediate enhanced immune-stimulatory effects through their heightened immunogenicity. In a breast cancer model, a recombinant fusogenic VSV variant carrying reovirus fusion-associated small transmembrane (FAST) proteins and the VSV(MΔ51) backbone (a more immunogenic variant of VSV) induced a significant increase in iNKT cells, NK cells, CD8
+ and CD4
+ T cells, and DCs in the tumor, compared to untreated and VSV-GFP-treated tumors [
30], which is consistent with our observations of immune cell responses to VSV-NDV treatment. This is likely due to the potent induction of ICD by the virus-mediated cell fusion [
14,
31].
A valid concern in the OV field is that viral vectors may induce strong anti-viral immune responses, which could counteract the therapy and detract from an efficient induction of anti-tumoral immunity. It was recently reported that oncolytic VSV-IFN-β led to the expansion of dominant antiviral effector CD8
+ T cells, which coincided with the timing of an observed reduction in anti-tumor T cell populations [
32]. Interestingly, in the model used here, we demonstrated a strong correlation between the presence of anti-viral and tumor-specific CD8
+ TILs, suggesting that VSV-NDV therapy not only does not distract from the induction of tumor-specific immunity, but it might even promote it. Notably, the activation of antiviral memory T cells has previously been associated with tumor growth inhibition, and viral peptides have been shown to sensitize B16 melanoma to PD-L1 blockade [
30]. Furthermore, prior immunization with NDV has been linked to superior tumor clearance and survival in mice [
31], underscoring the potential of antiviral immune responses to mediate potent anti-tumor effects, which seems to also hold true for VSV-NDV.
This study additionally revealed an early increase in innate immune cells, marked by the elevation of activated iNKT cells, NK cells, and DCs in TDLNs from the treated tumor, implicating their involvement in the initial immune response. The collective findings from numerous studies indicate that NK cells frequently impact favorably on the therapeutic outcomes of OV therapy [
33,
34,
35,
36,
37,
38]. This phenomenon can be explained by the pivotal role of the interaction between NK cells, T cells, and DCs. NK cells and T cells have the capability to recruit classical type 1 DCs (cDC1s) through the secretion of chemokines [
39], and multiple reports have documented the occurrence of the cross-priming of CD8
+ T cells by DCs in response to virotherapy [
40,
41,
42,
43]. It should be noted, however, that NK cells also play an important role in clearing oncolytic viruses, often limiting their oncolytic efficacy [
44]. Therefore, the dual role of NK cells, driving both antitumor responses as well as antiviral responses, must be carefully considered. Although a stronger innate immune cell response was expected within the virus-treated TME, our findings may be specific to the subcutaneous tumor setting of the applied model or to the timepoints investigated.
While it was beyond the scope of this work to characterize the precise mechanism and cross-talk among the individual immune cell types, a critical dependence on the cytotoxic T cell response for the therapeutic outcome of VSV-NDV therapy is strongly implicated by the complete ablation of tumor responses in the context of CD8+ T cell depletion. While the importance of CD8+ T cells in OV therapy is not completely unexpected, our results imply that the cytotoxic T cell response may even represent the more potent mechanism of action compared to the direct oncolytic effect. The fusion-mediated tumor lysis seems to be a critical driver of these T cell responses, as a concerted consequence of the therapeutic reprogramming of the TME that promotes immune cell infiltration and activation.
While these data provide a first proof-of-concept of the dynamic immune cell responses to this novel OV therapy, it is important to recognize the limitations of the B16OVA model. The artificial OVA antigen leads to potent immune responses that likely exceed those against natural TAAs, potentially leading to an overestimation of efficacy [
45,
46,
47,
48]. This model served as a preliminary screening tool to identify immune cell responses to VSV-NDV therapy, which will certainly need to be validated in more immunologically predictive models and compared across distinct tumor indications.