1. Introduction
Hazardous gases (such as CO
x, NO
x, and NH
3) produced by industrial activities and vehicles cause respiratory diseases in humans, contribute to the greenhouse effect, and damage the environment [
1]. For the safety of human health and environmental protection, gas sensor technology is crucial for the detection of these gases. Due to unique properties of nanomaterials, two-dimensional materials such as graphene, silicene, Mxene, phosphorene, and transition metal dichalcogenides have attracted significant interest in various studies [
2,
3,
4,
5,
6,
7,
8]. For gas sensing applications, graphene exhibits great performance due to its excellent mobility and high surface-to-volume ratio [
9,
10,
11]. However, the absence of a band gap in graphene makes it difficult to control charge in the sheet [
9,
12]. Borophene, a two-dimensional material composed of monolayer of boron atoms, has been successfully synthesized on silver surface under ultrahigh vacuum conditions [
13,
14]. Borophene has gained widespread attraction for research in nanotechnology because of its unique electronic, optical, thermal, magnetic, and mechanical properties [
15,
16,
17,
18,
19]. Boron possesses three valence electrons in 2s
22p
1 orbitals which can form sp
2 hybridization [
9,
15] and the bonding between boron atoms is highly complex with polymorphisms [
13]. This complexity leads to various physical and chemical properties [
20,
21], which distinguish it from other two-dimensional materials like graphene and transition metal dichalcogenides. Owing to a variety of atomic arrangements, there have been reports on theoretical and experimental studies of borophene structures with several stable structures including triangular and hexagonal planar or quasi-planar structure motifs [
17,
22,
23,
24,
25,
26]. An anionic
cluster (
) is a highly symmetric planar wheel-like structure of borophene composed of a single boron atom at the center of a
ring [
27,
28]. The structure provides double aromaticity and high electronic stability resulting from the bonding between the central boron atom and the B
8 ring through three delocalized σ and three delocalized π bonds [
27]. Doping with metal atoms can modify the structures and properties of borophene with small planar or quasi-planar structures tailored for specific applications [
27,
29,
30,
31,
32]. The structural diversity offers enormous potential to tailor the properties of borophene for applications in areas ranging from electronics [
33,
34], energy storage [
35,
36], gas sensing [
9,
37,
38], etc.
Focusing on gas sensing applications, the extraordinary geometries, high surface-to- volume ratio, and excellent electronic properties of borophene allow it to be used as gas sensors. Several theoretical studies have reported on the adsorption of gas molecules by borophene. For examples, Huang et al. [
37] calculated adsorption energies, charge transfer, and electronic structures of borophene with buckled and line-defective phases for adsorption of NH
3, NO, NO
2, and CO using density functional theory (DFT). Shukla et al. [
9] used DFT calculation combined with non-equilibrium Green’s function (NEGF) to study adsorption behavior of monolayer borophene for gas molecules of CO, CO
2, NO, NO
2, and NH
3. Ta et al. [
38] theoretically investigated the potential for the adsorption of five main hazardous gases including CO, NO, NH
3, NO
2, and CO
2 on β
12 borophene by using van der Waals (vdW) density functionals (vdW-DFs). These studies showed that borophene could be developed as sensing materials for gas sensors to detect toxic gases. However, the mentioned studies focused on gas adsorption of pristine borophene sheets with different structures.
Recently, borophene quantum dots (BQDs) have been explored for various applications, including solar cell [
39], biosensors [
40,
41], catalysts [
42], supercapacitors [
43], and biomedical applications [
44]. For instance, Liu et al. reported the preparation of BQDs/BC
2N heterostructures for an ultrasensitive humidity sensor. The sensor showed ultra-high sensitivity, high selectivity, fast response and recovery times, and good flexibility over a wide detection range (11–97% RH) by using the Grotthuss chain reaction sensing mechanism via hydrogen bonding and multilayer physisorption [
45]. The BQDs exhibit quantum confinement effects, where their electronic and optical properties are influenced by the size and shape of the dots [
40,
46]. However, research on the application of pristine and chemically functionalized BQDs for gas sensing application is still limited.
In the present study, we theoretically investigate the adsorption behaviors of NO2, CO2, CO, and NH3 gas molecules on wheel structures of pristine and nitrogen functionalized BQDs using self-consistent charge density functional tight-binding (SCC-DFTB) method. We examine adsorption energies, optimal adsorption sites, charge transfer, density of states (DOS), and electronic properties to understand the interaction between these gas molecules and the pristine and nitrogen functionalized BQDs.
2. Calculation Methods
The SCC-DFTB method is derived from second-order expansion of the DFT Kohn–Sham total energy in terms of the charge density fluctuations [
47,
48,
49]. The Kohn–Sham approach simplifies the complex problem of interacting electrons by mapping it onto a system of non-interacting electrons that reproduce the same electron density. Although the non-interacting system is easier to solve, it still provides the correct ground-state electron density for the original interacting system. In the DFTB approach, the traditional self-consistency between the effective potential and the charge density is replaced by a simpler self-consistency in the distribution of Mulliken charges. The method is optimized for high accuracy by using a minimal basis set to represent the single-electron Kohn–Sham-like eigenstates. This allows electronic interactions to be precomputed within a two-center model using an effective Hamiltonian derived from a carefully chosen reference density [
50]. Based on Kohn–Sham orbitals, the total energy for the SCC-DFTB method is expressed as [
51,
52]:
where is the Kohn–Sham orbitals, is unperturbed Hamiltonian, and are the induced charges on the atoms A and B, respectively, is the second derivative with respect to its total charges (Coulombic-like interaction potential), and denotes repulsive potential for the pair of atoms A and B.
The benchmark of the SCC-DFTB method is a balance between accuracy and computational efficiency with reduced computational costs compared to standard DFT [
47,
49]. For large systems, the SCC-DFTB calculations were 2–3 orders of magnitude faster than DFT [
47,
51]. It should be noted that that calculation time depends on both the number of atoms and the complexity of the structures being simulated. While the system in this work may not be considered large in absolute terms, SCC-DFTB still ensures faster computation times. The SCC-DFTB method simulates electronic properties, energies, and molecular structures of molecules and materials, and provides results in good agreement with DFT methods [
53]. For gas adsorption, the SCC-DFTB method can be used to investigate interaction between gas molecules and sensing materials [
54,
55,
56]. The benchmark of the SCC-DFTB method has been performed for theoretical studies of boron clusters [
52,
57].
In this work,
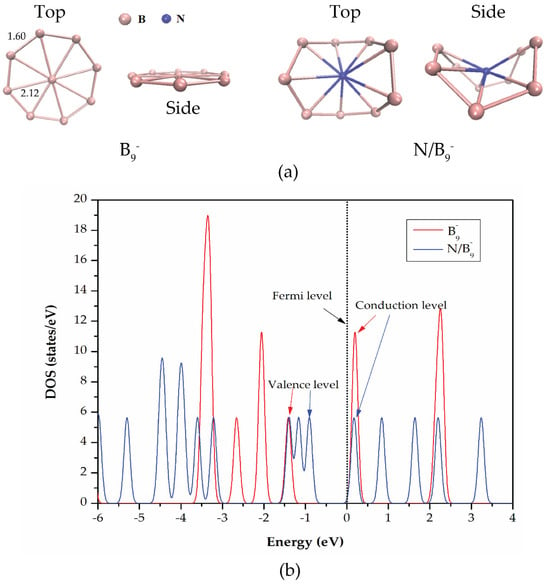
wheel-like structures of pristine and nitrogen functionalized BQDs (
and
BQDs) were built. For nitrogen functionalization, a nitrogen atom was at the center of the
ring and bonded with nine surrounding boron atoms. It should be noted that this configuration is theoretically feasible because nitrogen can bond to boron forming a stable structure. However, experimental implementation can be challenging due to the precision required to control the doping process and ensure that the nitrogen atom is properly positioned at the center of the B9 ring. Techniques like molecular beam epitaxy or chemical vapor deposition which allow for atomic-level precision may be required making the process more complex but achievable with advanced fabrication methods. To reduce the impact of edge effects, we applied periodic boundary conditions along with a large simulation box to investigate the QDs. This approach ensures that the QDs’ electronic structure, adsorption phenomena, and charge transfer effects are accurately accounted for providing more reliable insights compared to calculations using a single unit cell. The matsci-0-3 parameter set was used for calculation in O-N-C-B-H system. In addition, the matsci-0-3 parameter set well describes the interaction of covalent organic system, especially small organic molecules containing nitrogen, oxygen, carbon, and hydrogen [
49,
51,
58,
59]. The SCC-DFTB command code and its relative parameters are demonstrated in
Source Code S1 of Supporting Information. To investigate the most favorable adsorption sites, gas molecules including NO
2, CO
2, CO, and NH
3 were placed at different distances with orientation configurations (parallel and perpendicular) above the surfaces of
and
BQDs. Adsorption strengths between the structures and gas molecules are described in the term of adsorption energies (E
ad). The adsorption energy is calculated as the following equation:
where , and are the total energies of or BQDs with gas molecules, BQDs, and gas molecules, respectively. A negative value of Ead indicates that gas molecules are adsorbed and more negative values are more favorable adsorption sites.
To study charge transfers from/to gas molecules resulting from the adsorption, net charge transfer is defined as the charge difference of gas molecules before and after adsorption [
60]. The net charge transfer (Q) is obtained from Equation (3):
where and are the charges of gas molecules adsorbed on the surfaces of BQDs structures and isolated gas molecules, respectively.
Total density of states (DOS) was calculated to analyze the influence of gas molecule adsorption on the electronic properties of BQDs. The k-point sampling density was set to
for the DOS studies [
37].
4. Conclusions
In summary, we have studied the adsorption behavior of and BQDs structures for NO2, CO, CO2, and NH3 gas molecules using SCC-DFTB method. Structural geometries, the most favorable adsorption sites and electronic properties were investigated. After nitrogen functionalization, the structural and electronic properties of BQDs were changed. The structure was buckled and its energy gap was decreased due to the impurity states. Based on the adsorption results, the adsorption energies of the structures were found to be higher than those of borophene sheets. Additionally, the adsorption energies of the BQDs were higher than those of the BQDs. The BQDs favored NO2 adsorption while the BQDs favored CO2 with parallel orientation. The calculated charge transfer indicated that there was a transfer of charge from the BQDs structures to the gas molecules. In case of DOS analysis, new local states formed around the Fermi level after gas adsorption. According to high adsorption energies, DOS and interaction distances, the interaction between gas molecules and the and BQDs was found to be chemisorption. Our calculation suggests that nitrogen-doped BQDs are promising candidates for detecting hazardous gases in the environment. We believe that this work will provide valuable guidance on future studies in both simulations and experiments.