1. Introduction
Glaucoma, an optic neuropathy with the loss of retinal ganglion cells (RGCs), is a leading global cause of irreversible vision loss and blindness [
1]. The common critical pathological phenotypes of this optic neuropathy include progressive optic nerve axon degeneration and RGC death, ultimately with some efficacy, but they are often inadequate to stop disease progression [
1,
2]. Hence, investigating the underlying pathophysiological mechanisms of glaucoma is crucial. Understanding these mechanisms could supplement IOP-lowering strategies or offer independent approaches to improve treatment efficacy.
Vision impairment in common glaucomatous retinal degenerative conditions is significantly influenced by oxidative stress, a recognized risk factor in glaucoma [
3,
4,
5,
6,
7]. Therapeutic strategies targeting oxidative stress and mitochondrial dysfunction are of interest for potential glaucoma treatments. Importantly, clinical studies have linked primary open-angle glaucoma to specific mitochondrial cytochrome c oxidase subunit I of the oxidative phosphorylation (OXPHOS) complex IV [
8] and single-nucleotide polymorphisms of toll-like receptor 4 (TLR4) [
9]; this suggests a connection between OXPHOS stress-induced mitochondrial dysfunction and TLR4-mediated neuroinflammation in glaucoma neurodegeneration. However, the precise mechanisms linking oxidative stress, mitochondrial dysfunction, and neuroinflammation in glaucoma remain poorly understood.
Apolipoprotein A-I binding protein (AIBP; gene name APOA1BP alias also known as NAXE) is a secreted protein that regulates cholesterol removal from the plasma membrane [
10,
11,
12]. Extracellular AIBP binds to TLR4, thereby directing cholesterol depletion to inflammatory cells with high levels of TLR4 expression [
13,
14]. Intracellular AIBP is localized in mitochondria and modulates mitophagy by regulating Parkin and mitofusin (MFN) 1 and MFN2. Under oxidative stress conditions, this regulatory mechanism helps eliminate damaged mitochondria in macrophages [
13,
14,
15]. We have demonstrated that AIBP expression is significantly downregulated in mouse and human glaucomatous retina [
14,
15] and that AIBP deficiency induces mitochondrial dysfunction in Müller glial cells [
14,
15,
16]. Since Müller glial cells regulate retinal neuroinflammation [
14,
15,
16], we hypothesized that AIBP deficiency is associated with RGC damage and visual dysfunction under oxidative stress conditions.
In the present study, we found that AIBP deficiency exacerbated the oxidative stress-induced impairment of mitochondrial dynamics, biogenesis, and function in Müller glia, leading to impaired visual function. Conversely, the administration of recombinant AIBP (rAIBP) restored mitochondrial dynamics and function in the retina and improved visual function under oxidative stress. rAIBP administration reduced TLR4-associated lipid rafts, restored mitochondrial dynamics and function, decreased inflammasome activation and inflammatory response, and reduced Müller glial cell death. This protection may promote RGC survival and restore visual function by ameliorating glia-driven mitochondrial dysfunction and neuroinflammation in glaucoma.
2. Materials and Methods
2.1. Animals
C57BL/6J (The Jackson Laboratory, Bar Harbor, ME, USA) and AIBP knock-out (
Apoa1bp−/−) mice were housed in covered cages, fed with a standard rodent diet ad libitum, and kept on a 12 h light/12 h dark cycle. C57BL/6J mice were bred in-house for experiments and used as wild-type (WT) mice.
Apoa1bp−/− mice on a C57BL/6J background were generated in our laboratory, as previously reported [
15]. Animals were assigned randomly to experimental and control groups. Visual function tests were studied with 10-month-old male and female mice. Research in ophthalmic vision involving animals was conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic Vision Research and under protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Diego (CA, USA). (IACUC S12063 for mouse).
2.2. Recombinant AIBP
N-terminal His-tagged AIBP was produced in a baculovirus/insect cell expression system to allow for post-translational modification and to ensure endotoxin-free preparation as previously described [
17,
18]. A bulk production of recombinant AIBP was ordered from Selvita Inc. (Kraków, Poland) and was stored at −80 °C.
2.3. Induction of Retinal Oxidative Stress
To induce oxidative stress, mice received intraperitoneal (IP) injections of paraquat (PQ) (15 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) in saline solution three times for 1 week as previously described [
19]. Measurements for visual function tests (
n = 6 to 8 mice per group) such as pattern electroretinogram (pERG), pattern visual evoked potential (pVEP), and virtual optomotor response were assessed 1 week after PQ treatment.
2.4. Tissue Preparation
Mice were anesthetized by an IP injection of a mixture of ketamine (100 mg/kg, Ketaset; Fort Dodge Animal Health, Fort Dodge, IA, USA) and xylazine (9 mg/kg, TranquiVed; Vedeco, Inc., St. Joseph, MO, USA) before cervical dislocation. For immunohistochemistry (n = 3 mice per group), the retinas were dissected from the choroids and fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS (pH 7.4) for a duration of 2 h at 4 °C. Retinas were washed several times with PBS then dehydrated through graded levels of ethanol and embedded in polyester wax. For Western blot and PCR analysis, extracted retinas were immediately used.
2.5. Immunohistochemistry
The immunohistochemical staining of 7 μm wax sections of full-thickness retinas was performed. Sections from wax blocks from each group (
n = 4 retinas/group) were used for immunohistochemical analysis. To prevent non-specific background, tissues were incubated in 1% bovine serum albumin (BSA, Sigma-Aldrich)/PBS for 1 h at room temperature before incubation with the primary antibodies for 16 h at 4 °C. After several wash steps, the tissues were incubated with the secondary antibodies for 4 h at 4 °C and subsequently washed with PBS. The sections were counterstained with the nucleic acid stain Hoechst 33342 (1 μg/mL; Invitrogen, Carlsbad, CA, USA) in PBS. Images were acquired using Keyence All-in-One Fluorescence microscopy (BZ-X810, Keyence Corp. of America, Itasca, IL, USA). Each target protein fluorescent integrated intensity in pixel per area was measured using the ImageJ software version 1.54i [National Institutes of Health (NIH), Bethesda, MD, USA]. All imaging parameters remained the same and were corrected with background subtraction. The primary and secondary antibodies used in this study are presented in
Supplementary Table S1.
2.6. pERG and pVEP Analysis
pERG and pVEP were measured with a Celeris apparatus (Diagnosys, Lowell, MA, USA) as previously reported [
14,
15]. After dark adaptation overnight, the mice were anesthetized with intraperitoneal injections of a cocktail of ketamine/xylazine under red light, and a mixture of 0.5% tropicamide and 2.5% phenylephrine was applied directly to the eye to dilate pupils. Eyes were also treated with 1% proparacaine drops and 0.3% hypromellose gel to prevent corneal drying and cataracts. pERG responses were recorded using alternating, reversing, black and white vertical stimuli at 1 Hz (2 reversals per second) and 50 candela/m
2 delivered by the pattern stimulator. Then, 200 traces were recorded per eye, and averaged waveforms were calculated in which amplitudes (µV) were measured from the P1 peak to the N2 trough. At the same time, pVEP responses were recorded. For all the recordings, ground and reference needle electrodes were placed subcutaneously in the tail and snout, and the active electrode was placed subdermally in the midline of the head at the location of the visual cortex. Each eye was separately exposed to 100 flashes of 1 Hz, 0.05 cd s/m
2 white light through the corneal stimulators and recorded for 300 ms with a sample frequency of 2000 Hz. Then, 200 traces were recorded per eye, and averaged waveforms were calculated in which amplitudes (µV) were measured from the P1 peak to the N1 trough. For each mouse, we performed five trials and swept 100 times per trial. The low- and high-filter frequency cutoffs for pVEP were set to 1.25 Hz and 100 Hz. All data measurements were recorded while maintaining a constant body temperature between 37 °C and 38 °C using the system’s heat pads. The data were analyzed by the software Espion V6 (Diagnosys) [
20,
21].
2.7. Virtual Optomotor Response Analysis
Spatial frequency was analyzed on a virtual optomotor system (OptoMotry; Cerebral Mechanics Inc., Lethbridge, AB, Canada) as previously reported [
15,
19,
22]. Unanesthetized mice were placed on an unrestricted platform in the center of a virtual cylinder comprising four monitors arranged in a square (arena) that project a sinusoidal grating (i.e., white versus black vertical bars) rotating at 12 deg/s. Mice were monitored by a camera mounted at the top of the arena, while a cursor placed on the forehead centered the rotation of the cylinder at the animal’s viewing position. To assess visual acuity, tracking was determined when the mouse stopped moving its body and only head-tracking movement was observed. The spatial frequency threshold, a measure of visual acuity, was determined automatically using the accompanying OptoMotry software (Version 2.1.0), which uses a step-wise paradigm based upon head-tracking movements at 100% contrast. Spatial frequency began at 0.042 cyc/deg, which gradually increased until head movement was no longer observed.
2.8. Cell Culture and rAIBP Administration
rMC-1 cells, immortalized rat retinal Müller glial cell line (Kerafast, Boston, MA, USA), were grown in Dulbecco’s Modified Eagle Medium (DMEM, Corning Inc, New York, NY, USA) supplemented with 5% fetal bovine serum and 1% penicillin/streptomycin solution at 37 °C in a humidified CO2 incubator. The cells were seeded into six-well plates at a density of 2 × 105 cells/well and maintained for 24 h. Subsequently, the cells were pretreated with 0.2 μg/mL rAIBP or BSA as a control for 2 h, followed by stimulation with PQ (500 μM) for 24 h. A cell culture experiment was performed with 3 independent experiments.
2.9. siRNA Transfection
rMC-1 cells were transfected with scramble siRNA or AIBP siRNA purchased from Origene (Rockville, MD, USA) using the Amaxa Nucleofector (Lonza Group, Basel, Switzerland). Briefly, the cells (3 × 106) were resuspended in 100 μL of nucleofector solution mix followed by the addition of 20 nM of scramble siRNA or AIBP siRNA and transfected according to the manufacturer’s instructions. The cells were seeded into six-well plates at a density of 5 × 105 cells/well and maintained for 24 h.
2.10. Mitochondrial Membrane Potential (MMP) and Mitochondrial Reactive Oxygen Species (mtROS) Measurement
rMC-1 cells were seeded into 6-well plates at a density of 2 × 105 cells/well, maintained for 24 h, and treated with the indicated materials for 24 h. MMP was determined by flow cytometry. The cells were incubated with TMRE solution (200 nM, Invitrogen) for 30 min at 37 °C. The intracellular mtROS level in rMC-1 cells was measured by flow cytometry. After treatment, the cells were incubated with mitoSOX solution (500 nM, Invitrogen) at 37 °C for 30 min.
2.11. Western Blot Analyses
Retina tissues and rMC-1 cells were homogenized on ice for 1 min using a modified RIPA lysis buffer [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate (SDS)], containing protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, San Diego, CA, USA), and incubated on ice for 30 min for complete cell lysis. Harvested retinas were homogenized in RIPA buffer using a motorized tissue grinder (ThermoFisher Scientific). Cell and tissue debris was removed by centrifugation at 12,000×
g for 15 min. Lysates (10 μg of protein) were separated by 4–20% Mini-PROTEAN TGX-precast protein gel electrophoresis (Bio-Rad, Hercules, CA, USA), and target protein levels were determined by Western blot analysis [
19]. The membranes were blocked with 5% non-fat dry milk in PBS/0.1% Tween-20 (PBS-T) for 1 h at room temperature, then incubated with primary antibodies overnight at 4 °C. After washing several times with PBS-T, the membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse or rabbit IgG (1:1000–7000; Bio-Rad; Cat# 1721011 or 1706515) for 1 h at room temperature and developed using an enhanced chemiluminescence substrate system. The images were captured and quantified using the ImageQuant™ LAS 4000 system (GE Healthcare Bio-Science, Piscataway, NJ, USA) and ImageJ software (NIH). For each experiment, we conducted multiple biological replicates (at least 3 replicates), quantified the results, calculated averages, and presented the data as bar graphs. The blots shown were carefully selected based on these quantitative data to ensure clarity and consistency.
2.12. Immunocytochemistry
rMC-1 cells were fixed in 4% paraformaldehyde for 15 min at room temperature. After gently washing, the cells were permeabilized with 0.1% triton X-100 and incubated with a cleaved caspase-3 antibody for 16 h at 4 °C. After three wash steps, the cells were incubated with Alexa Fluor-568 conjugated donkey anti-rabbit IgG antibody for 2 h. For nuclear staining, the cells were further incubated with Hoechst 33342 (1 μg/mL, Invitrogen) for 5 min. Images were acquired using Keyence All-in-One Fluorescence microscopy (BZ-X810, Keyence). Each target protein fluorescent integrated intensity in pixel per area was measured using the ImageJ software (NIH) as described above.
2.13. Quantitative Real-Time PCR (qRT-PCR) Analysis
Total mRNAs were isolated from rMC-1 using a Trizol reagent. cDNAs were prepared from 1 μg of RNA using a SuperScript III First-strand synthesis system (Invitrogen). qRT-PCR was performed with the PowerUp SYBR master mix (Applied Biosystems, Forster City, MA, USA) according to the manufacturer’s instructions. The mRNA levels of target genes were determined and quantitated using their specific primers and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [
23]. The primers used in this study are presented in
Supplementary Table S2.
2.14. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Analyses
rMC-1 cells (5 × 10
4 per well) were seeded into Seahorse XF24-well plates. At 24 h, cells were pretreated with either BSA or rAIBP (0.2 μg/mL) for 2 h, followed by exposure to PQ (50 μM). OCR and ECAR were measured using an XF24 Extracellular Flux analyzer (Agilent, La Jolla, CA, USA). For OCR analysis, we used a Seahorse XF cell mito stress test kit (Agilent) as previously reported [
5,
24,
25]. After measuring basal respiration, oligomycin (2 μg/mL, Sigma-Aldrich), an inhibitor of ATP synthesis; carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 μM, Sigma-Aldrich), the uncoupler; and rotenone (2 μM, Sigma-Aldrich), an inhibitor of mitochondrial complex I, were sequentially added to measure maximal respiration, ATP-linked respiration, and spare respiratory capacity. For ECAR analysis, we used a Seahorse XF cell glycolysis stress test kit (Agilent). Glucose (10 mM, Sigma-Aldrich), oligomycin (1 μM, Sigma-Aldrich), and 2-deoxyglucose (2-DG, 50 mM, Sigma-Aldrich) were sequentially added to measure glycolysis, glycolytic capacity, and glycolytic reserve.
2.15. Immunofluorescence Staining of TLR4-Associated Lipid Rafts
After exposure to oxidative stress, rMC-1 cells were immediately put on ice, washed once with cold PBS, and fixed with 4% PFA for 10 min. Cells were washed twice with cold PBS and incubated with blocking buffer containing 5% FBS for 30 min without permeabilization, followed by staining with Cholera Toxin B (CTxB)-Alexa Fluor 594 (Invitrogen) to stain lipid rafts and rabbit anti-TLR4 antibody (Proteintech, Rosemont, IL, USA) for 2 h at room temperature, washed and incubated with anti-rabbit Alexa Fluor 647 conjugated secondary antibody (Invitrogen) for 1 h at room temperature. Cells were washed five times, and coverslips were mounted with Prolong Gold with DAPI (Invitrogen) into slides. Image acquisition was conducted using Keyence All-in-One Fluorescence microscopy (BZ-X810, Keyence), and image analysis was performed using ImageJ software (NIH). A colocalization assessment was executed using the colocalization finder plugin, facilitating the calculation of Pearson’s coefficients.
2.16. Statistical Analysis
For comparison between two groups that have a small number of samples related to a fixed control, statistical analysis was conducted utilizing nonparametric analysis and a one-sample t-test. For comparison between two independent groups, a two-tailed Student’s t-test was performed. For multiple group comparisons, we used either a one-way ANOVA or two-way ANOVA, using GraphPad Prism (Version 10, GraphPad, San Diego, CA, USA). Statistically, significance was defined as a p value below 0.05.
4. Discussion
Our recent study showed a crucial link between Müller glial cells and the increased expression of TLR4 and IL-1β in glaucomatous retinas [
14,
15,
16]. In addition, we found that administering rAIBP protects RGCs against acute IOP elevation by suppressing cytokine production and inflammatory response [
15]. Nevertheless, the precise mechanisms through which AIBP confers protection in Müller glial cells and their mitochondria have yet to be fully elucidated. This study unveils significant new findings suggesting that AIBP protects Müller glial cells against oxidative stress. In AIBP-deficient mice, the detrimental effects of oxidative stress on mitochondrial function and inflammatory reactions are exacerbated, leading to worsening vision impairment. Notably, the administration of rAIBP mitigates these consequences by reducing TLR4-associated lipid rafts. This action preserves mitochondrial dynamics and function while dampening inflammatory responses in Müller glial cells under oxidative stress, ultimately improving RGC and visual function.
Müller glial cells, the most abundant glial cells in the retina, exhibit a radial arrangement that spans the entire retinal thickness. Müller glial cells play crucial roles in the retina by establishing vital connections with retinal neurons. These functions encompass maintaining cholesterol balance, clearing waste products through phagocytosis, shielding neurons from excessive exposure to neurotransmitters like glutamate, and supplying end products from anaerobic metabolism [
32,
33,
34,
35]. Reactive glial cells, such as astrocytes and microglial cells, are closely associated with glaucomatous neuroinflammation [
36]. In contrast, the involvement of reactive Müller glial cells and their mitochondrial dysfunction remains to be elucidated in glaucomatous neuroinflammation. Oxidative stress, mitochondrial dysfunction, and inflammation associated with glial activation are crucial pathogenic mechanisms in the glaucomatous retina [
5,
15,
36,
37]. Our recent findings indicated that Müller glial cells lacking AIBP display extensive mitochondrial fragmentation and reduced ATP production, resulting in Müller glial cell dysfunction linked to oxidative stress and inflammatory response [
15]. Furthermore, our recent findings revealed a notable decrease in AIBP levels within glaucomatous human RGCs and Müller glial cells [
14,
15]. This is accompanied by increased cholesterol accumulation and TLR4-associated lipid raft formation, as well as MAPK activation, metabolic energy stress, and inflammatory responses in Müller glial cells [
14,
16]. Hence, these findings strongly suggested that oxidative stress could be a critical process regulated by AIBP in the context of retinal inflammation and associated with mitochondrial stress in Müller glial cells in glaucomatous neuroinflammation.
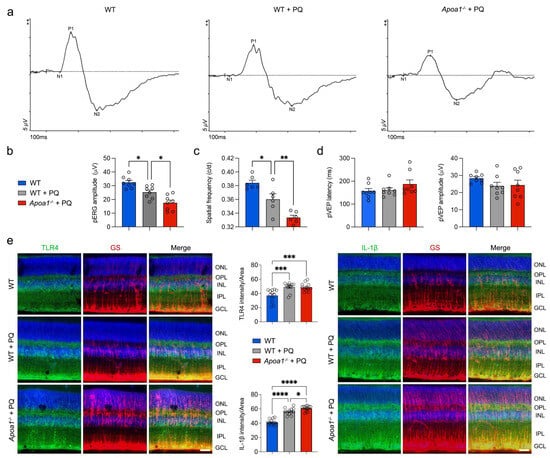
In this study, we observed a deterioration in visual function in
Apoa1bp−/− mice when exposed to oxidative stress, correlating with exacerbated impairments in mitochondrial dynamics, OXPHOS activity, and mitochondrial biogenesis in the
Apoa1bp−/− retina. Notably, these effects were associated with the activation of Müller glial cells, culminating in heightened inflammatory responses. Significantly, Müller glial cells lacking AIBP demonstrated increased vulnerability to degeneration, characterized by MMP loss, elevated mtROS production, excessive mitochondrial fragmentation, and compromised mitochondrial respiration, particularly under oxidative stress conditions. Oxidative stress is linked to the activation of multiple signaling pathways of MAPKs, such as p38 and ERK1/2 [
38,
39]. Since AIBP deficiency and glaucomatous damage activated p38 and ERK1/2 by increasing the phosphorylation of p38 and ERK1/2 in the retina [
14,
15], we observed that oxidative stress worsened the increase in the phosphorylation of p38 and ERK1/2 in Müller glial cells lacking AIBP. Thus, these findings strongly suggest a vicious cycle where oxidative stress initiates AIBP deficiency, leading to a cascade involving oxidative stress, AIBP deficiency, mitochondrial dysfunction, and MAPK activation in Müller glial cells. Ultimately, this cascade may trigger inflammasome activation, apoptotic cell death, and inflammatory response in oxidative stress-mediated glaucomatous neuroinflammation.
Both the restoration of AIBP expression and the administration of rAIBP significantly reduced neuroinflammation and protected RGCs in acute or chronic experimental animal models of glaucoma [
14,
15]. Recent evidence indicates that AIBP plays a multifaceted role, involving extracellular cholesterol efflux from the cell membrane and various intracellular functions within the mitochondria [
5,
11,
13,
15,
16,
40]. Specifically, intracellular AIBP has been shown to regulate autophagy/mitophagy in macrophages [
13]. This study suggested that mitochondria-associated AIBP enhances mitophagy, thereby contributing to mitochondrial quality control and preventing macrophage death in atherosclerosis [
13]. In this study, our findings indicated that administering rAIBP improved visual function in mice and maintained mitochondrial dynamics and function in retinal cells, including Müller glial cells, under oxidative stress conditions. These effects were supported by preventing mitochondrial fragmentation and restoring mitochondrial fusion activity and OXPHOS activity. Thus, our findings suggest that AIBP would be critical in protecting retinal mitochondria against oxidative stress. Further studies will delve into how administering rAIBP regulates mitophagy, thereby preserving mitochondrial quality control and safeguarding Müller glial cells.
Glia-driven neuroinflammation is evident in both glaucomatous human and mouse retinas, characterized by an increased expression of TLR4 and IL-1β within activated Müller glial cells [
14,
15]. Our observations are particularly noteworthy given that TLR4 activation typically triggers the MAPK/NFκB pathway and NLRP3 inflammasomes, resulting in an increased production of proinflammatory cytokines [
41,
42,
43]. We found that the administration of rAIBP notably diminished TLR4-associated lipid rafts in Müller glial cells subjected to oxidative stress. Furthermore, this effect was strongly correlated with reduced MAPK activation, a suppressed NLRP3-associated inflammasome pathway, and mitigated inflammatory responses in Müller glial cells under oxidative stress conditions. Importantly, considering the established role of AIBP in reducing cholesterol deposition in glaucomatous retinas [
14], our findings collectively suggest that extracellular rAIBP administration may potentially contribute to the prevention of neuroinflammation and cell death in Müller glial cells. This could be achieved through the inhibition of cholesterol deposition, reducing TLR4 inflammaraft formation, and the mitigation of mitochondrial dysfunction during oxidative stress-induced glaucomatous neuroinflammation and neurodegeneration.
Our study proposes a novel concept that oxidative stress triggers AIBP deficiency in Müller glial cells, which consequently increases the activation of TLR4 lipid rafts. This then leads to mitochondrial dysfunction, inflammasome activation, neuroinflammation, cell death, and, ultimately, vision impairment (
Supplementary Figure S6). Notably, our findings support the notion that administering rAIBP counteracts these dysfunctional outcomes, safeguarding Müller glial cells. Hence, this protection may promote RGC survival and restore visual function by ameliorating glia-driven mitochondrial dysfunction and neuroinflammation in glaucoma.