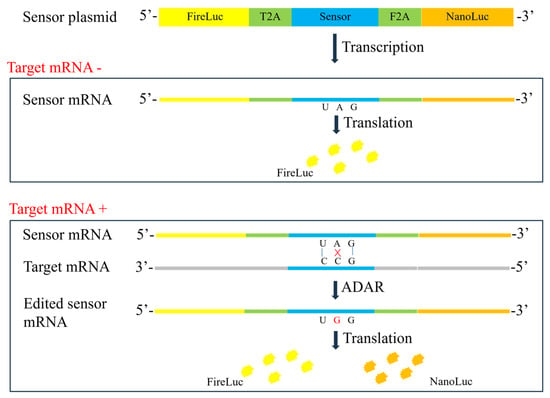
The expression level of therapeutic genes serves as an indicator of bioactivity in the quality control of gene therapy products. However, it is often difficult to quantify the protein expression of the therapeutic gene owing to a lack of specific antibodies or high background in the test cells [
12]. Although mRNA detection is more accessible, it is tedious [
26,
27,
28]. In the present study, an RNA editing-based reporter assay was developed to detect specific mRNA. The designed sensor RNA specifically identified the target mRNA, and the reporter gene was activated in a dose-dependent manner with the help of ADAR proteins. All sensors that targeted different regions, including SMN (GOI), GFP (tag sequence), and WPRE (3′UTR), exhibited a dose-dependent pattern. The SMN and GFP sensors contained the TAG stop codon. Once the internal A was converted to I (read as G), the translation of the downstream nano-luciferase was activated. However, no satisfactory CCA sequence was found for the WPRE sequence, necessitating the use of TCA. The WPRE sensor contained the TAA stop codon. Only when AA was converted to II (read as GG) was the translation of the downstream nano-luciferase activated. Of note, a high level of translational readthrough was observed in the sensor reporter, which was unexpected. As shown in
Figure 5, the nano-luciferase-to-firefly luciferase ratio was approximately 0.05 for the SMN and GFP sensors and 0.4 for the WPRE sensor, which are not in line with previous reports that suggested that the termination efficiency of TAA is better than that of TAG [
29,
30]. For the WPRE sensor, although the activation of the nano-luciferase required the conversion of two adenosines to inosines, its response (SNR) was not inferior to that of the SMN and GFP sensors, suggesting that the editing efficiency for AA is similar to that for A. To reduce the background luciferase activity, two tandem stop codons were introduced into the sensor sequence to increase the efficiency of translation termination. Thus, SMN dsensor 1 containing a single stop codon was used as a control. Meanwhile, SMN dsensor 1 + 2 contained two consecutive stop codons, and SMN dsensor 1 + 3 contained two stop codons separated by a 15-nucleotide sequence. As shown in
Figure 2d, the three sensors showed similar luciferase background activity, suggesting that there are other mechanisms responsible for the background activity of nano-luciferase. As expected, the A-to-G editing efficiency of SMN dsensor 1 was the highest. However, surprisingly, the A-to-G editing efficiency of SMN dsensor 1 + 2 was lower than that of dsensor 1 + 3. The reason for this phenomenon may be that the binding of the ADAR protein to one A shields another nearby A (not AA), thereby reducing its accessibility for editing. These findings indicate that the distance between two As has a significant impact on the process of coediting. Previous reports have shown that ADAR1 and ADAR2 overexpression can increase RNA editing efficiency [
23,
31]. A mutant ADAR2 (E488Q) has been shown to exhibit a higher RNA editing capacity [
32,
33]. In the current study, different ADARs, including ADAR1, ADAR2, and mutant ADAR2 (E488Q), were cotransfected. The results showed that overexpressed ADARs resulted in a higher luciferase background. However, no improvement in assay sensitivity and SNR was observed, suggesting that endogenous ADARs are sufficient for the mRNA reporter assay. Additional experiments are needed to verify whether other mechanisms besides ADAR-mediated RNA editing are involved in the activation of nano-luciferase, although it is clear from the specificity validation data that the activation is induced by a specific target sequence. Many attempts were made to optimize the assay, such as AAV receptor overexpression and cell pretreatment with hydroxyurea, but only prolonged incubation time could effectively increase the sensitivity and SNR of the assay.
After confirming the response of the sensor-based reporter to plasmids, other types of gene therapy vectors, including mRNA, lentivirus, and AAV, were analyzed. All these vectors exhibited a dose-dependent effect on specific sensor reporter cells. For AAV vectors, in particular, the sensor reporter assay could discriminate between different serotypes in terms of transduction efficiency, indicating that it can be employed as a potency assay for AAV products. Next, the suitability of the assay to determine the transcriptional activity of rAAV products was assessed. To verify the specificity of the sensors, AAV samples harboring different elements were evaluated using SMN, GFP, and WPRE sensor reporter 293T cells. The results revealed that each sensor responded only to the AAV sample containing its target sequence, suggesting high specificity. As a quantitative method, 4-PL parallel analysis is widely employed in cell-based potency assays [
34,
35,
36]. The developed reporter assay fit the 4-PL model, with an R
2 of >0.99. Considering that high AAV titer has a negative effect on the cell function, resulting in reduced firefly luciferase activity, which is also observed in lentivirus, the nano-luciferase-to-firefly luciferase ratio (reflecting the efficiency of RNA editing) was used as the response. F-tests revealed no significant nonlinearity or nonparallelism (
p = 0.05), indicating that the 4-PL parallel analysis is suitable for the reporter assay. The precision, accuracy, and linearity of the reporter assay were not inferior to conventional cell-based bioassays [
37,
38,
39,
40]. Of note, the linear range of the assay was very wide, and test samples with lower activity (such as AAV2-GFP-WPRE sample 2) gave precise and accurate results. Overall, the developed reporter assay showed a similar dose–response relationship to the protein assay (GFP expression), suggesting that it can be used as an alternative method for the potency testing of gene therapy products. Furthermore, its high precision, accuracy, and linearity made it suitable for the batch release and stability analyses of gene therapy products. The development of such a reporter assay only requires the screening and design of a target-specific sensor sequence, followed by its substitution for the existing sensor sequence. This streamlined approach greatly simplifies the method development process compared with existing assays.
Limitations of this study and future technical developments: The present study is based on the 293T cell line, which is known for its high susceptibility to AAV infection and inherent expression of ADARs. For other cell types, including those derived from neural tissue, overexpressed ADARs may be essential to increase the sensitivity of the assay. Furthermore, the assay is designed to target mRNA, and although we have established a correlation between mRNA and protein expression, this relationship may not be consistent in all biological contexts. This limitation suggests that further validation of the assay’s performance in different cell types and under different conditions is required. Finally, the underlying mechanism of the assay should be investigated to verify whether there is another mechanism other than ADAR-mediated mRNA editing, which will be of great importance for further improvements in assay performance.
Source link
Lei Yu www.mdpi.com