
2.1. Monoclonal Antibodies
Monoclonal antibodies (mAbs) were among the pioneering types of immunotherapy approved for anti-cancer treatment and hold a vital and increasingly important role in current treatment regimens. Notably, rituximab, the anti-CD20 mAb was the first mAb implemented in oncology and remains the most commonly applied [28]. The approval of rituximab in 1997 revolutionized the treatment of B-cell lymphomas and was an important milestone in the advent of newer immunotherapeutic approaches.
Rituximab is a chimeric human–mouse mAb that targets CD20, a common B-cell surface marker, and facilitates the specific depletion of these cells [29]. The CD20 antigen is highly expressed on the surface of B cells as well as the majority of B-cell lymphomas but is absent from hematopoietic stem cells, differentiated plasma cells, and other healthy tissues, making it a suitable target for the efficient induction of effector mechanisms that mediate the specific depletion of B cells [30]. Antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity are key mechanisms contributing to rituximab-mediated B-cell depletion [31]. Since mature plasma cells and B-cell progenitors do not express CD20, targeting and depleting B cells at these intermediate stages of development typically does not result in lasting side effects [32,33]. With over 20 years of clinical experience using rituximab, it has proven to be highly effective and safe, despite initial doubts. Rituximab has significantly improved outcomes in B-cell malignancies, including follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) [34,35]. Moreover, its high efficiency in B-cell depletion was found to be of central interest in the treatment of EBV-PTLD in the post-allogeneic hematopoietic stem-cell transplantation (alloHSCT) setting [36,37].
Based on this pioneering success of rituximab in clinical practice, over the last decade, we can observe the high expansion of novel mAbs approved in the hemato-oncology field, as summarized in Table 1. Ofatumumab is a second-generation, fully humanized anti-CD20 mAb that targets an alternative epitope than rituximab and binds its target with higher affinity. It was approved by the FDA for treating CLL refractory to fludarabine and alemtuzumab in 2009 as monotherapy, and later in 2014, for use in combination with chlorambucil [38,39]. However, it was discontinued in all non-US markets in 2019 due to economic reasons, and its use in those regions is restricted to compassionate access only. Nevertheless, it was recently approved with a new indication in treating relapsing forms of multiple sclerosis in adults [40]. Obinutuzumab, another second-generation anti-CD20 mAb, was approved by the FDA in 2013 for use with chlorambucil in CLL treatment and in 2016 for use with bendamustine in treating relapsed or refractory FL (R/R FL) [41,42]. Obinutuzumab (GA101) is a humanized, glycoengineered IgG1 type II mAb targeting the epitope on the large extracellular loop of CD20, which partially overlaps with the rituximab epitope [43]. Recent findings from a phase III trial have shown the superior effectivity of obinutuzumab over rituximab when included in immunochemotherapy for first-line treatment of FL [44]. Conversely, no advantage over rituximab was demonstrated in DLBCL [45]. However, the matter of the doses applied of both mAbs in comparable studies is open to discussion [35]. Several other clinical trials with obinutuzumab are in progress, including those comparing its effectivity over rituximab in other CD20-positive malignancies, like B-cell acute lymphoblastic leukemia (B-ALL) (NCT04920968) and mantle cell lymphoma [46,47,48]. Moreover, the first rituximab biosimilars, CT-P10 (Truxima®; developed by Celltrion) and GP2013 (Rixathon®; developed by Sandoz), were approved by the EMA in 2017 [49].
In addition to CD20, novel antigenic targets have been explored lately, and CD19, CD22, CD33, CD38, and SLAMF7 belong to the group most extensively studied in HM applications [50]. Elotuzumab, an anti-SLAMF7 mAb, was the first mAb approved in the treatment of MM and established a paradigm shift in immunotherapies for MM [51]. It demonstrated significantly improved 1-year and 2-year progression-free survival (PFS) rates of 68% and 41%, respectively, when combined with lenalidomide and dexamethasone, compared to these agents alone, achieving an overall response rate (ORR) of 79% [27]. It was approved by the FDA in 2015 in combination with lenalidomide and dexamethasone for the R/R MM treatment. New elotuzumab-containing combinations are in clinical trials [52].
CD38 is a transmembrane glycoprotein widely expressed on MM cells, and other mAbs targeting this attractive antigen are in active research [53]. Daratumumab, an anti-CD38 mAb, received FDA approval for treating relapsed or refractory multiple myeloma (R/R MM) in 2016, in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with MM who have received at least one prior therapy [54]. Another CD38 mAb, isatuximab was approved in 2020, in combination with pomalidomide and dexamethasone for the treatment of adult patients with MM who have received more than two prior therapies, and, in 2021, for combination with carfilzomib and dexamethasone, for the treatment of adult patients with R/R MM who have received one to three prior lines of therapy [55,56]. Moreover, in September 2024, the FDA approved isatuximab with bortezomib, lenalidomide, and dexamethasone for adults with newly diagnosed multiple myeloma who are not eligible for autoHSCT, based on results from the phase III IMROZ trial [57].
The latest approval of tafasitamab (Monjuvi®), a humanized, anti-CD19, Fc-modified mAb, was embraced enthusiastically among clinicians. DLBCL is the most prevalent form of non-Hodgkin lymphoma, comprising 25–45% of new lymphoma cases annually [26]. The introduction of rituximab in combination with cyclophosphamide, doxorubicin, prednisone, and vincristine (R-CHOP) as the standard first-line immunotherapy has significantly improved patient outcomes with this entity. However, 30–40% of patients still experience relapse or are resistant to this initial treatment [58]. For these relapsed or refractory patients, effective and well-tolerated alternative treatments remain scarce, resulting in a poor prognosis. On 31 July 2020, the FDA granted an accelerated approval for Monjuvi® in combination with lenalidomide for adult patients with R/R DLBCL who are not eligible for autologous stem-cell transplant [59]. The approval was granted based on findings from the single-arm, phase II L-MIND study (NCT02399085), which enrolled 81 participants. Patients were treated with tafasitamab at a dose of 12 mg/kg intravenously in combination with lenalidomide for up to twelve 28-day cycles, followed by tafasitamab monotherapy. The primary endpoint was to assess the best ORR and duration of response. Among the 80 patients who received the tafasitamab–lenalidomide combination, 60% achieved an objective response, 43% had a complete response, and 18% had a partial response. In 71 DLBCL patients, the best ORR was 55%, with 37% achieving complete and 18% partial responses. The median duration of response was 21.7 months [60,61,62,63]. Numerous novel mAbs are being investigated in clinical trials and the marketing authorization of mAbs is an actively growing field, nicely reviewed annually by the “Antibodies to Watch” series [9,10].
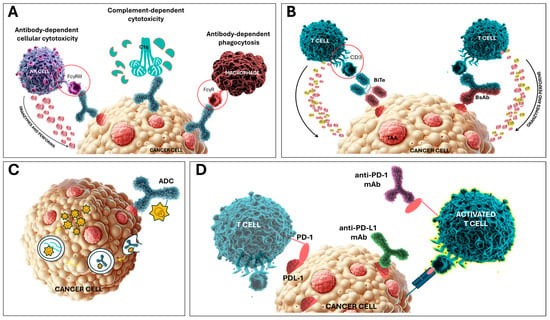
2.2. Bispecific Antibodies and T-Cell Engagers
The improvement of mAb-based immunotherapy brought about the idea of bispecific Abs (BsAbs), which can bind two different target antigens or two different epitopes on the same antigen simultaneously [64,65]. Various designs of BsAbs have been developed over time differing in format and size [66]. The most widely used BsAb format is based on selective recruitment of potent effector cells of the immune system toward tumor cells. This is achieved by simultaneously binding a tumor-associated antigen (TAA) on tumor cells, and an activating receptor on immune effector cells, such as a CD3 molecule on the effector T lymphocytes in the case of the class of T-cell engagers [67,68]. Such BsAb-mediated cross-linkage results in the activation of the effector cells in a non-MHC-restricted manner, leading to efficient tumor cell eradication [69]. Another discrimination factor within bispecifics is the presence or absence of an Fc region. The interaction between Fc domains and their receptors on various types of immune effector cells such as natural killer (NK) cells, monocytes, and macrophages, is capable of inducing antibody-dependent cell-mediated cytotoxicity. The Fc domain can also bind complement to elicit complement-dependent cytotoxicity, leading to the unnecessary non-specific immune response during bsAb treatment [70]. Hence, the shorter fragments (without Fc portions) are currently more explored, usually consisting of single-chain variable regions (scFvs) of two parenting mAbs connected by a flexible linker [71,72]. Of these novel bispecific platforms, the bispecific T-cell engagers (BiTEs) have demonstrated the greatest clinical relevance so far, with Blinatumomab being the standout example [73].
Blinatumomab is a CD19xCD3 BiTE approved for the treatment of patients with Philadelphia chromosome-negative precursor B-cell acute lymphoblastic leukemia (B-ALL) [74]. Due to its small size (~55 kDa), blinatumomab can reach within close proximity to a T cell and target-cell epitopes, which enables highly specific T-cell activation [75]. The human CD19 antigen is a transmembrane glycoprotein from the immunoglobulin superfamily. It is specifically expressed on normal and neoplastic B cells, as well as on follicular dendritic cells [76]. The surface expression of CD19 begins during B-cell lymphopoiesis, around the time of immunoglobulin gene rearrangement, and its expression is tightly regulated throughout B-cell development and maturation, ceasing during terminal plasma cell differentiation [77]. In mature B cells, CD19 expression is three times higher than in immature B cells. It plays a critical role in regulating B-cell signaling thresholds by modulating both B-cell receptor-dependent and independent pathways [78]. The fact that CD19 is expressed by a wide range of B-lymphoid malignancies, but not by hematopoietic stem cells and pro-B cells, makes it an attractive target for antibody-mediated therapy [79]. Blinatumomab has demonstrated significant efficacy in adult and pediatric patients with R/R B-ALL and patients with measurable residual disease (MRD). Clinical studies showed that over 70% of MRD-positive patients achieved MRD negativity, which correlates with better long-term outcomes and improved overall survival (OS) [80]. Based on its acceptable safety profile and high response rates, blinatumomab was approved by the FDA for treating R/R B-ALL in both adult and pediatric patients in 2014 [73]. Since then, the amount of clinical data on blinatumomab has continuously increased. The NEUF retrospective observational study investigated the safety and effectiveness of blinatumomab in adult patients with R/R B-ALL in real-world clinical settings. A total of 140 R/R B-ALL patients were included, with 106 being Ph− and 34 Ph+. These real-world data support the efficacy results seen in randomized clinical trials of blinatumomab [81]. Moreover, recent data show the encouraging efficacy of blinatumomab in certain ALL contexts, including its use as a frontline therapy, as a bridge to transplantation, and in “chemotherapy-free” combination treatment regimens, both in adults and pediatric populations [82,83,84,85]. In a phase III randomized clinical trial, pediatric patients with high-risk, first-relapse B-ALL received blinatumomab as a consolidation therapy before undergoing alloHSCT. This treatment led to improved event-free survival (EFS) and higher rates of minimal residual disease remission compared to chemotherapy. The EFS benefit was seen across all patient subgroups, including those with extramedullary disease and very early relapse (within 18 months) [86]. A longer follow-up revealed significantly better OS in patients treated with blinatumomab compared to chemotherapy, regardless of their MRD status before treatment [87]. Furthermore, the latest post hoc analysis of this study shows that children with high-risk, first-relapse B-ALL who received blinatumomab as the third consolidation therapy before alloHSCT had improved 2-year OS and EFS estimates compared to those who received HC3 (dexamethasone, vincristine, daunorubicin, methotrexate, ifosfamide, and PEG-asparaginase), regardless of whether they were treated with total body irradiation plus etoposide or chemoconditioning [88]. However, the sample size and the post hoc nature of the analysis may have limited applicability, hence the ongoing FORUM study, which includes over 1700 registered children with ALL, is expected to provide more comprehensive data on the benefits of using blinatumomab before alloHSCT in real-world settings [89].
Building on its successful outcomes, additional BsAbs have been developed over time and studied in clinical trials, but we have only seen a real breakthrough in the last two years. Six novel BsAbs for hemato-oncology applications achieved FDA approval in the last two years, as summarized in Table 2. The CD20xCD3 BsAbs mosunetuzumab, epcoritamab, and glofitamab have been granted accelerated FDA approval for certain subtypes of non-Hodgkin lymphoma due to their significant efficacy in clinical trials [90].
BCMA is currently the main target for immunotherapies in MM due to its highly restricted expression, minimizing the potential of off-target effects. It is predominantly expressed on differentiated plasma cells and plasmablasts, while not expressed on naïve B cells, hematopoietic stem cells, or normal non-hematologic tissues [91,92]. Teclistamab, approved in 2022, is a novel T-cell-redirecting BsAb targeting BCMA and CD3. It was evaluated in the phase I/II MajesTEC trial involving heavily pretreated patients with R/R MM [93]. The ORR achieved 65%, with a median PFS of 11.3 months [94]. Another BsAb-targeting BCMA and CD3 is elranatamab. It was approved in 2023 for adult patients with R/R MM who have undergone at least four prior therapies, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 mAb, showing promising results in clinical trials [95]. In the MagnetisMM-1 trial, involving heavily pretreated R/R MM patients, the median PFS was 11.8 months, and the OS was 21.2 months. At a 12-month follow-up, the ORR was 63.6%, with 38.2% achieving a CR. Cytopenias and cytokine release syndrome (CRS) were reported, though no dose-limiting toxicities occurred. In the phase II MagnetisMM-3 trial, patients responding to weekly elranatamab were transitioned to bi-weekly dosing to enhance tolerability, resulting in a 61.0% ORR and a 35% CR rate. At 15 months, the duration of response (DOR) rate was 71.5%, PFS was 50.9%, and OS was 56.7%, with infections, CRS, anemia, and neutropenia being common adverse events [96,97,98].
BsAbs targeting myeloma cell antigens beyond BCMA have demonstrated potential in heavily pretreated R/R MM patients, including those who have previously received BCMA-targeted treatments [99]. The unique example in this class is talquetamab. Besides binding CD3 on T cells with one arm, the second one specifically binds to G-protein-coupled receptor family C, group 5, member D (GPRC5D), an orphan receptor present on the surface of normal plasma cells and myeloma cell lines [100]. In the MonumenTAL-1 phase I study, talquetamab showed promising results in heavily pretreated R/R MM patients, with an ORR of 70% for intravenous and 64% for subcutaneous administration, and a median DOR of 10.2 and 7.8 months, respectively. CRS was reported in 77–80% of patients. Follow-up data confirmed a sustained ORR of 73% with a median DOR over one year. In the phase II MonumenTAL-2 study, the combination of talquetamab and pomalidomide demonstrated efficacy and tolerability in the same patient population [101,102].
2.3. Antibody–Drug Conjugates
Immunoconjugates, or antibody–drug conjugates (ADCs), are a group of mAbs attached to a given cytotoxic or radioactive payload working as sophisticated delivery systems. The specific binding of Ab with its antigen enhances the targeted delivery to the tumor site, increasing the efficacy of the small molecule while reducing side effects and non-specific toxicity to non-target tissues [103]. The Ab can also be linked to a radionuclide to more precise targeted radiotherapy at the tumor site [104]. The initial enthusiasm for these targeted drug delivery systems surged with the approval of gemtuzumab ozogamicin in 2000 but waned following its withdrawal, due to significant hepatotoxicity in 2010 [105]. However, updated data on the clinical effectiveness and safety of gemtuzumab ozogamicin given in a fractionated dosing regimen resulted in its re-approval in 2017 for both newly diagnosed and R/R acute myeloid leukemia (AML) cases [106]. Technological advancements, including a better selection of cytotoxic agents and the use of smaller conjugates, have significantly increased the potential clinical benefits of ADCs [107]. Several ADCs have been designed and applied for clinical use in hematologic malignancies and their targets include CD19, CD20, CD22, CD30, CD33, CD37, CD79, CD123, and BCMA [108,109].
Currently, there are thirteen FDA-approved ADCs in total, and seven of them with hemato-oncology applications (as summarized in Table 3). In addition, more than 100 ADC molecules are at different stages of clinical trials.
Brentuximab vedotin (BV), an anti-CD30 Ab conjugated to the microtubule inhibitor monomethyl auristatin E (MMAE), has demonstrated efficacy in treating relapsed/refractory Hodgkin lymphoma (R/R HL) and systemic anaplastic large-cell lymphoma. These data led to its FDA approval for cancer treatment in 2011 and for post-autologous HSCT consolidation in 2015 [110]. In 2018, based on results from the phase III ECHELON-1 clinical trial, the FDA expanded the approval of brentuximab vedotin to include the treatment of adult patients with previously untreated stage III or IV classical Hodgkin lymphoma (cHL) in combination with AVD (doxorubicin, vinblastine, and dacarbazine) [111]. Lately, the update after 5 years of follow-up was published, in which BV+AVD (brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine) demonstrated significant and lasting improvements in PFS compared to ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine), regardless of PET-2 status, while maintaining a consistent safety profile [112]. Furthermore, the BV was tested in clinical trials with newly diagnosed advanced-stage cHL led by the German Hodgkin Study Group, investigating its use in combination with chemotherapy. In the randomized, multicenter, open-label, phase III, HD21 clinical trial the BrECADD regimen (BV, etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone) was compared with the escalated BEACOPP regimen (bleomycin, etoposide, doxorubicin, vincristine, prednisone, and procarbazine) in a PET2-guided treatment approach. The BrECADD regimen demonstrated superior PFS compared to eBEACOPP, with 4-year PFS rates of 94.3% versus 90.9%, respectively. OS rates were similar between the two regimens, at approximately 98.5%. Further, BrECADD showed a more favorable safety profile, with significantly fewer treatment-related morbidity events (42% vs. 59%). It also preserved gonadal function better than eBEACOPP, with higher recovery rates of fertility markers in both men and women. These findings suggest BrECADD is more effective and better tolerated, positioning it as a strong alternative to eBEACOPP for advanced-stage classical Hodgkin lymphoma [113].
In 2017, inotuzumab ozogamicin (INO), a humanized anti-CD22 mAb linked to the cytotoxic antibiotic calicheamicin, was approved as a monotherapy for the treatment of CD22-positive B-ALL [114]. Within the INO-VATE trial, inotuzumab ozogamicin demonstrated significantly superior efficacy compared to the standard of care (SC) in patients with R/R B-ALL. The CR and CR with incomplete hematologic recovery (CRi) rates in the first 218 randomized patients, were 80.7% in the inotuzumab ozogamicin group versus 29.4% in the SC group [115]. Additionally, patients receiving INO achieved significantly higher rates of MRD negativity, longer PFS, and improved 2-year OS in an ad hoc analysis [114,115]. Furthermore, the analysis of pooled data from INO-VATE and the earlier phase I/II “Study1010” by Marks et al. revealed that salvage treatment with INO can provide a bridge to transplant in R/R B-ALL, and patients treated with INO were approximately one-third less likely to experience a relapse compared to those who received SC [116]. Hence, the role of INO as the bridge to HSCT is currently intensively investigated [117,118,119]. However, careful consideration is necessary when applying inotuzumab ozogamicin before the myeloablative conditioning regimen in HSCT recipients, due to unique adverse events, including multi-organ failure and a significant risk of veno-occlusive disease [120,121]. The encouraging data on the effectiveness of INO paved the way for studies investigating its use in first-line treatment alone or combined with reduced-intensity chemotherapy or other targeted therapies [122]. These clinical trials have predominantly targeted older adults, as they tend to experience poorer outcomes historically, often due to their limited tolerance to intensive chemotherapy, the frequent presence of high-risk disease characteristics, and reduced eligibility for HSCT [123]. The German Multicenter Study Group on Adult Acute Lymphoblastic Leukemia (GMALL) conducted the INITIAL-1 trial, an open-label phase II study, to evaluate the efficacy and safety of INO as an induction therapy for older adults with newly diagnosed Ph- B-ALL, the results of which were published recently [124]. Participants received up to three cycles of INO as induction therapy, followed by standard chemotherapy-based consolidation and maintenance treatments. The study demonstrated high remission rates, with all 43 patients achieving either CR or CRi, primarily after the first induction cycle, and MRD negativity improving to 74% by the third cycle. Survival outcomes were promising, with OS rates of 91% at one year and 81% at two years, and EFS at one year reaching 88%. The treatment was associated with common grade 3 or higher adverse events, including leukocytopenia, anemia, thrombocytopenia, and elevated liver enzymes. Only one case of suspected veno-occlusive disease occurred, highlighting a manageable safety profile for the therapy. These findings suggest that incorporating INO into induction regimens may improve outcomes for this patient population.
Polatuzumab vedotin (PV) is an mAb targeting CD79b delivering monomethyl auristatin E (MMAE), an anti-mitotic toxin, to cancer cells. In June 2019, the FDA granted accelerated approval for polatuzumab vedotin, in combination with bendamustine and rituximab, for treating adults with relapsed or refractory DLBCL who have undergone at least two prior therapies [125]. CD79b, a component of the B-cell receptor complex, is crucial for downstream signaling and is expressed in the majority of B-cell lymphomas, making it a promising immunotherapeutic target [126]. The approval of polatuzumab vedotin is a considerable advancement in treatment options for patients with R/R DLBCL, particularly for those ineligible for transplantation or CAR-T therapy [127]. Ongoing trials will determine whether bendamustine–rituximab chemoimmunotherapy is the optimal partner for polatuzumab in treating DLBCL patients. Early studies of various combinations have demonstrated antitumor activity and a manageable safety profile in R/R FL and frontline non-Hodgkin lymphoma settings [128,129]. Lately, we have observed a paradigm shift in the frontline treatment of DLBCL thanks to the POLLARIX clinical trial which challenged the current standard—the R-CHOP regimen [130]. In this trial, 879 newly diagnosed patients were randomly assigned to receive either the R-CHOP or PV-R-CHP regimen, in which vincristine was replaced by PV. After a median follow-up of 28.2 months, PFS was higher in the PV (76.7% compared to 70.2% in the R-CHOP group), although no significant differences in OS at the two-year mark were observed. Moreover, the safety profile of the novel PV regimen was similar to that in the R-CHOP arm. These results led to the FDA approving PV as a first-line treatment for adult patients who have previously untreated diffuse large B-cell lymphoma, not otherwise specified or high-grade B-cell lymphoma, and who have an International Prognostic Index score of 2 or greater, marking the first significant update to initial therapy for DLBCL in two decades [131].
Belantamab mafodotin (BelaMaf) was the only ADC approved for MM treatment, although other ADCs are under investigation. This ADC molecule is an mAb specific to BCMA and linked to the toxin auristatin F. Its use was intended for patients who have received at least four prior therapies, including an anti-CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent [132]. The DREAMM-1 and DREAMM-2 studies demonstrated its effectiveness as a single agent in patients with advanced MM, achieving high responses (ORR of 32% and 35%) and long-term remission durations of approximately one year [133]. However, the phase III randomized DREAMM-3 trial, which aimed to evaluate the safety and efficacy of BelaMaf as a single agent compared to the PomDex (Pomalidomide and dexamethasone) regimen in patients with R/R MM, failed to show a PFS benefit [134]. These results were a base to withdraw BelaMaf by the FDA in November 2022 and subsequently, the conditional marketing authorization for Blenrep has not been renewed by the European Commission, as of October 2024. Nevertheless, the collected clinical data do not undermine completely the effectiveness of BelaMaf. Hence, many combination therapies are being tested to improve its safety and effectiveness profile. Notably, the latest results from the DREAMM-7 and DREAMM-8 studies are highly encouraging.
The DREAMM-7, phase III, multicenter, open-label, randomized study aimed to evaluate the efficacy and safety of BelaMaf in combination with bortezomib and dexamethasone (BorDex) compared to a combination of daratumumab and BorDex in patients with R/R MM. Results published this year indicate that the BelaMaf combination nearly tripled the median PFS compared to the standard daratumumab combination (36.6 months vs. 13.4 months), with a 59% reduction in the risk of disease progression or death [135]. These findings suggest that the BelaMaf plus BorDex regimen may offer a superior treatment option for R/R MM patients compared to the current standard of care.
The DREAMM-8 study, in turn, compared BelaMaf combined with pomalidomide and dexamethasone vs. pomalidomide plus bortezomib and dexamethasone (PVd) in R/R MM patients after one prior line of therapy. Similarly, the BelaMaf-based combination demonstrated superior performance compared to the control group in response rate, response durability, and PFS [136]. This combination significantly extended the time to disease progression or death compared to the PVd regimen. At a median follow-up of 21.8 months, the median PFS was not yet reached in the BelaMaf group, whereas it was 12.7 months in the PVd group. The ORR was higher in the BelaMaf arm, 77% compared to 72% in the PVd group. Notably, the CR was 40% in the BelaMaf group versus 16% in the PVd group. Among patients achieving CR or better, the MRD negativity rate was 23.9% in the BelaMaf group, compared to 4.8% in the PVd group, indicating deeper responses to the BelaMaf regimen. The safety and tolerability of this regimen were consistent with the known profiles of the individual agents. These results suggest that the BPd combination offers a significant improvement in progression-free survival and depth of response for patients with relapsed or refractory multiple myeloma, potentially redefining treatment standards in this setting. Additionally, a few other combination therapies are being tested to improve its effectiveness profile [137].
Loncastuximab tesirine consists of a CD19-targeting mAb linked to a pyrrolobenzodiazepine (PBD) dimer cytotoxic agent [138]. After binding to the CD19 antigen on B cells, the ADC is internalized, releasing the PBD dimer, which induces DNA cross-linking and prevents cancer cell replication, ultimately leading to cell death. The FDA approved loncastuximab tesirine in April 2021 for the treatment of R/R DLBCL, including DLBCL arising from low-grade lymphoma, in patients who have received at least two prior systemic therapies. In clinical trials, loncastuximab tesirine demonstrated significant efficacy in heavily pretreated patients, with durable responses, particularly in those with aggressive forms of DLBCL. A phase I study of loncastuximab tesirine in patients with R/R B-cell non-Hodgkin lymphoma (NHL) demonstrated an ORR of 45.6% in evaluable patients, with 26.7% achieving CR. Specifically, the ORR was 42.3% in patients with DLBCL, 46.7% in those with mantle cell lymphoma (MCL), and 78.6% in patients with FL [139].
Additionally, a multicenter, open-label, single-arm phase II trial (LOTIS-2) was conducted in patients with R/R DLBCL who had received two or more prior multiagent systemic treatments. The trial reported an ORR of 48.3%, a CR rate of 24.1%, and an OS of 9.9 months [140]. The results of LOTIS-1 and LOTIS-2 showed that loncastuximab tesirine had significant single-agent antitumor activity and durable response in R/R DLBCL patients, with acceptable safety and tolerability. Several clinical trials are ongoing to assess its safety and efficacy in NHL in various clinical settings, alone or in combination with other therapeutics [141].
2.4. Immune Checkpoint Inhibitors
Recognizing that neoplasm cells can hijack immune checkpoint pathways like cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death-1 (PD-1) to evade the immune system, immune checkpoint blockade therapy was developed as a novel immunotherapeutic approach [142]. Physiologically, immune checkpoints serve crucial functions in self-tolerance to avoid auto-immunity and tightly regulate the effector functions of activated T cells [143,144]. Immune checkpoint inhibitors (ICIs) are a class of mAbs that work by blocking immune checkpoint proteins from binding with partner molecules, thereby releasing immune breaks [145]. This strategy has proven effective against several solid tumors, including melanoma, non-small-cell lung cancer, renal cell carcinoma, and urothelial cancer, so far [146]. Over the past decade, various ICI drugs have received FDA approval including anti-CTLA-4 (ipilimumab), anti-PD-1 (pembrolizumab, nivolumab, and cemiplimab), and anti-PD-L1 (atezolizumab, avelumab, and durvalumab). However, their efficacy in HMs remains under extensive investigation. At present, the only FDA-approved ICI therapeutics are for classic Hodgkin lymphoma and primary mediastinal B-cell lymphoma. As knowledge of cancer biology and the tumor microenvironment (TME) expands, it is becoming evident that disease-specific factors, including the immune landscape of the disease, heavily shape both resistance and response to ICIs [147].
Classic Hodgkin lymphoma (cHL) has distinct biological features, marked by the presence of malignant Hodgkin and Reed–Sternberg (HRS) cells, which make up about 2% of the tumor cell population. The rest of its TME largely consists of reactive T cells and other immune cells, although these reactive immune cells exhibit an exhausted phenotype. This is caused mainly because of the over-expression of PDL-1 and PDL-2 on the malignant HRS cells, resulting in a loss of effector function of immune cells [148,149]. Hence, given its biology, the immune checkpoint blockade was first investigated in R/R cHL.
The first ICI tested in cHL was nivolumab, an anti-PD-1 mAb. The pivotal CheckMate-205 phase II trial reported an ORR of 69%, with 16% CR in 243 patients with R/R cHL following the failure of autoHSCT. The median time to response was approximately two months, and the median duration of response was 16.6 months [150]. The 5-year follow-up of the study demonstrated favorable OS and confirmed the efficacy and safety of nivolumab in R/R cHL after auto-HSCT failure [151]. Based on the results from the CheckMate-205 and CheckMate 039 trials, the FDA granted accelerated approval to nivolumab in 2016 for patients with R/R cHL who had failed autoHSCT and brentuximab vedotin treatment [152,153]. In addition, a breakthrough was achieved in the study comparing the combination regimen of nivolumab+AVD (doxorubicin, vinblastine, and dacarbazine) with current standard brentuximab+AVD in patients with advanced newly diagnosed Hodgkin’s lymphoma [154]. This multicenter, open-label, randomized trial enrolled 994 patients at least 12 years of age with stage III or IV previously untreated classic Hodgkin lymphoma. The outcomes of the study demonstrated significantly improved PFS in the nivolumab arm compared with the brentuximab vedotin arm (94% vs. 86% 1-year PFS rate). Additionally, the repeated analysis after one more year of follow-up revealed that the 2-year PFS rates were 92% in the nivolumab arm vs. 83% in the brentuximab vedotin arm. The 2-year EFS rate was 90% in the N+AVD arm and 81% in the BV+AVD arm. Although longer follow-up data are needed, this novel combination poses a promising option to revolutionize the treatment approach for elderly patients with advanced-stage cHL.
Pembrolizumab, another anti-PD-1 inhibitor, approved for use in melanoma, non-small-cell lung cancer, and head and neck cancers, has also been evaluated in HL treatment. In two clinical trials (KEYNOTE013 and KEYNOTE087), pembrolizumab has demonstrated significant efficacy in HL, with ORR ranging from 65% to 85%, depending on the cohort [155,156]. Furthermore, a phase III randomized controlled trial, Keynote-204, which compared pembrolizumab to brentuximab vedotin (BV) in the R/R setting, demonstrated an enhanced median PFS with pembrolizumab (13.2 months) compared to BV (8.3 months), with better tolerance to pembrolizumab vs. BV [157]. Based on these studies, the FDA granted accelerated approval to pembrolizumab for the treatment of cHL patients who are refractory or have relapsed after three or more prior therapies [158].
Additional studies with pembrolizumab are currently underway, focused on identifying the optimal combinations and timing for applying these agents in HL, and other lymphomas. Nevertheless, existing evidence highlights the remarkable responsiveness of HL to immune checkpoint blockade therapies [154,155,156,157,158]. The PD-1 blockade has also been investigated as a consolidation treatment following autoHSCT in high-risk patients with relapsed cHL, showing improved PFS, but more data are being awaited for optimal evaluation of these schemes [159,160,161].
Besides HL, PD-1 blockade has demonstrated significant efficacy in primary mediastinal and gray zone lymphomas, and Richter’s transformation of CLL [162,163,164,165,166]. Other lymphomas, particularly DLBCL in relapsed transplant-ineligible patients or as post-transplant consolidation, and FL, have shown limited benefit, as reviewed by others in [167,168]. ICIs have also provided minimal benefit in MM in studies performed so far, whether used alone or in combination with immunomodulatory agents like lenalidomide [169,170,171].
Source link
Justyna Jureczek www.mdpi.com