
1. Introduction
Rodents and shrews constitute two large orders of mammals that contain diverse species with widespread geographic distribution and ecology. These small animals have been established as reservoirs for a range of viruses and are linked to many human diseases. Hantaviruses and arenaviruses are two groups of zoonotic viruses of concern that are harbored by rodents and cause severe viral hemorrhagic fever. Both hantaviruses and arenaviruses are known to cause chronic infections in rodents, which shed the virus in urine, feces, and saliva. Humans can then become infected by contact with contaminated surfaces and desiccated excreta. Since the discovery of Lassa virus in 1969, no novel hemorrhagic fever-associated arenaviruses have been reported from Africa until Lujo virus (LUJV) was identified in 2008 [1,2]. It was discovered in South Africa during a nosocomial outbreak and transmission of human disease with high case fatality [3]. Despite multiple efforts, the reservoir of the virus has not been determined. Previous attempts to identify a reservoir for Lujo virus had shown the circulation of several arena- and paramyxoviruses in Zambian murine and shrew species [4,5,6]. The 2014 Zambian study analyzed over 400 wild rodents and 31 wild shrews collected across four locations in Zambia and reported various paramyxoviruses from different species of rodents. Paramyxoviruses are large, enveloped RNA viruses with a negative-sense, non-segmented genome [7]. The identified viruses were related to members of the paramyxovirus genera Morbillivirus, Narmovirus, and Henipavirus and the Tailam, Beilong, and J viruses of the genus Jeilongvirus. Jeilongviruses have been observed in rodents in Asia and Australia but were later also reported in rodents in Africa [8,9,10].
Similarly, knowledge about the prevalence, ecology, and phylogeny of adenoviruses, parvoviruses, or picornaviruses in rodents and shrews from Africa is limited. Adenoviruses are non-enveloped, icosahedral DNA viruses with a linear, double-strand genome [11]. They infect humans and animal species with infections ranging from asymptomatic to mild to severe fatal diseases. Adenoviruses are mostly considered to be species-specific, but cross-species transmission is possible, and serological findings suggest wider host ranges for some adenoviruses, e.g., canine adenovirus may also infect wolves, walruses, black and polar bears, and fishers [12,13,14]. There are currently three recognized murine adenovirus species: Murine mastadenovirus A (represented by murine adenovirus 1; MAdV-1), Murine mastadenovirus B (murine adenovirus 2; MAdV-2), and Murine mastadenovirus C (murine adenovirus 3; MAdV-3). Between them, MAdV-2 is divergent from the more closely related MAdV-1 and -3 [15,16]. Circulation of adenoviruses in multiple African wild rodent species has been reported in three studies that indicated a relationship to MAdV-2 based on short, PCR-generated sequences [17,18,19].
Parvoviruses are small, non-enveloped DNA viruses with a linear single-strand genome that cause disease in humans and other animal species [20]. Pathogenic mouse kidney parvovirus (MKPV) has been described in laboratory mouse strains [21,22,23,24], and murine chapparvovirus (MuCPV) in free-ranging urban house mice (Mus musculus) with undetermined pathology [25]; both are viruses of the species Chaphamaparvovirus rodent1 in the genus Chaphamaparvovirus. From wild rodents there is only an additional ~1kb NS1 sequence from a house mouse in China reported [26], and we are not aware of reports from wild rodents in Africa.
Aichiviruses are small, non-enveloped, icosahedral RNA viruses with a positive-sense, non-segmented genome and are classified into several species in the genus Kobuvirus [27]. Human Aichi virus (aichivirus A1, species Aichivirus A) is separated into three genotypes, A–C, that can cause gastroenteritis and other forms of enteric disease, particularly in children or immuno-compromised individuals [28,29] The virus is transmitted by the fecal–oral route and frequently reported from wastewater or contaminated foods, especially shellfish. The species comprises additional viruses from canines, felines, birds, and rodents (currently nine ‘types’, aichivirus A2 to A10), including murine kobuviruses (A3/A8), rat kobuviruses (A6/A9), vole kobuvirus (A7), and bat kobuvirus (A10). Although there are some reports on kobuviruses in rodents from Asia, the USA, and Europe, we are not aware of any on rodents in Africa [25,30,31,32,33,34,35,36,37,38].
We analyzed tissues from rodents trapped in the Katete district of eastern Zambia using a novel capture sequencing assay that has the potential for more efficient virus detection and discovery compared to prior approaches. VirCapSeq-VERT is a sample and pathogen-agnostic approach shown to detect all vertebrate viruses as well as novel viruses not known at the time of assay design [39]. VirCapSeq-VERT uses a set of 1 million biotinylated oligonucleotides to enrich for viral sequences [40]. These biotinylated capture oligonucleotides are hybridized to conventionally prepared sequencing libraries, then trapped by magnetic streptavidin beads, washed, and the thus virus-enriched material is finally subjected to high-throughput sequencing.
4. Discussion
Rodents and shrews frequently come into contact with humans. It is critical to assess viral diversity and identify novel viruses circulating within such reservoir species. Rodent surveillance in Zambia and southern Africa is important, as exemplified by the still-unknown reservoir of Lujo virus. Although we did not find Lujo virus, our non-targeted approach (VirCapSeq-VERT) generated data that substantially extend our knowledge about circulating rodent viruses in Zambia and serve as a model for routine reservoir species surveillance for potential future zoonotic transmission events [48]. As strengthened by recent SARS-CoV, SARS-CoV-2, or influenza A virus H5N1 emergences, proper pre-pandemic surveillance must not only include exotic or novel viruses but also all animal species in direct and frequent contact with humans and/or production livestock.
Numerous examples of zoonotic transmission of hanta-, arena-, pox-, and other viruses exist. Prior publications have, for example, noted the presence of picorna-like calhevirus in the enteric virome of shrews [49]. Calheviruses have also been found in human stool [50], and while it is plausible that detection of viruses such as calhevirus in human stool is an incidental finding (due to consumption of food containing the virus), adaptation to new hosts upon continued viral exposure is possible so that monitoring in humans and contact animals appears prudent.
Our study showed aichivirus sequences in the organs of multimammate mice that represent a novel strain in the species Aichivirus A. Presence in organs with a substantial prevalence (11%) is more compatible with infection than incidental presence in non-sterile sites, e.g., in rectal swabs or feces via ingestion [29,51]. However, little is known regarding the pathology and tissue tropism of rodent aichiviruses. Previous studies have been performed mainly on fecal samples [25,30,31,33,34,35,36,37,38,52,53], and only four studies report tissue samples [25,32,35,36]. No overt pathology has been reported in any of the studies, as expected for reservoir hosts that are commonly not significantly affected by the infection. A potential for cross-species transmission of these viruses has been discussed based on phylogenetic analyses [30,35,54,55]. In ongoing VirCapSeq-VERT studies in Zambia of pediatric respiratory disease, pneumonia cases, and pediatric diarrhea cases, we found related aichivirus sequences in nasopharyngeal and stool samples (unpublished data). Human Aichi virus sequences have been found previously in Africa, in diseased children from Tunisia (genotype A), Ethiopia (no genotype reported), Burkina Faso (genotypes A, B, and C), and a single case from Nigeria (genotype B) [52,53,56,57]. Aichi virus has also been reported from environmental samples in Tunisia (genotypes A and B) and South Africa (no genotype reported) [53,58,59]. To our knowledge, this is the first report of an aichivirus A from African rodents, and it indicates the multimammate mouse as a prominent host species in Zambia. Considering the wide host species distribution—the species Aichivirus A currently includes viruses from humans, dogs, cats, mice, rats, bats, and birds—our data support a potential risk for zoonotic transmission.
The family Paramyxoviridae includes several important human pathogens, such as measles (genus Morbillivirus, subfamily Orthoparamyxovirinae) and mumps viruses (genus Orthorubulavirus, subfamily Rubulavirinae), as well as classical zoonotic viruses transmitted from fruit bat reservoirs, like Nipah or Hendra virus (both members of the genus Henipavirus, subfamily Orthoparamyxovirinae) [60]. Bats became an increasing focus of research after the SARS and MERS outbreaks and, more recently, the COVID-19 pandemic. However, rodents represent more zoonotic agents, have a larger diversity of species than bats, and are a prominent reservoir for viruses with pandemic potential [61,62]. Jeilongviruses form a young but rapidly growing genus in the subfamily Orthoparamyxovirinae that currently combines viruses found in rodents, cats, bats, shrews, hedgehogs, and tenrecs. Most of them share features such as an enlarged G ORF, accessory ORFs SH and/or TM, and where studied, a unique V protein-independent inhibition of signal transducer and activator of transcription (STAT) translocation in response to interferon activation [63]. Although the zoonotic potential of jeilongviruses remains unknown, some insights into biology and pathogenicity have been gained through studies on J-virus. J-virus can cause fatal disease in mice, and serosurveys have shown antibodies in mice, rats, pigs, a cow, and humans. However, experimental infection of pigs did not result in clinical disease, while neutralizing antibodies did develop in some infected animals [64,65]. Different isolates of J-virus exist; strain LW is not pathogenic in mice, and strain BH causes severe disease. Sequence analyses of recombinants indicated a role in J-virus pathogenesis for three single base mutations in which the two strains differ [66]. It has also been shown that the SH gene product is involved in pathogenicity; removal of SH in recombinant J-virus BH led to increased tumor necrosis factor alpha (TNF-α) production and apoptosis in vitro and to attenuation in mouse infections, similar to SH protein function in human mumps virus [67,68,69]. Milanzi virus includes an SH ORF, possibly linked to immune evasion and pathogenicity. Lupande virus matches previously reported short, PCR-derived L-gene sequences from Zambia and lacks the SH ORF. Combined with the previous study in Zambia [5], our data indicated that Lupande virus is widespread and highly prevalent. Lupande virus and the related memana virus [10] were found in the same mouse species, although in two distant regions of Africa (Zambia and Guinea). Of known jeilongviruses with the same genome organization and from countries neighboring Zambia, Ruloma virus from Tanzania [9] and Mount Mabu Lophuromys virus 1 and 2 from Mozambique [8] were found in the same rodent species, Lophuromys machangui, but only Ruloma virus maps in the same clade as Lupande virus (see Figure 3A). Milanzi virus branches in a monophyletic clade together with mainly short, PCR-derived sequences from Tunisia, Madagascar, and Reunion [70,71]. The clade also includes longer sequences from Asia, in part with gaps so that the overall genome organization remains unknown for some [72,73]. The current evolutionary grouping focused on the L gene sequence may not be optimal, as it does not consider pathogenicity factors (e.g., SH) and/or genes related to host specificity (G or F) [74,75,76]. We therefore favor a more differentiated, staged approach that takes additional genes and genome organization into account. Conserved L gene sequence may be appropriate for distant relationships at the family or subfamily level, but additional information from other genome regions may be added for genus demarcation criteria and full coding sequence considered for resolution at the species level.
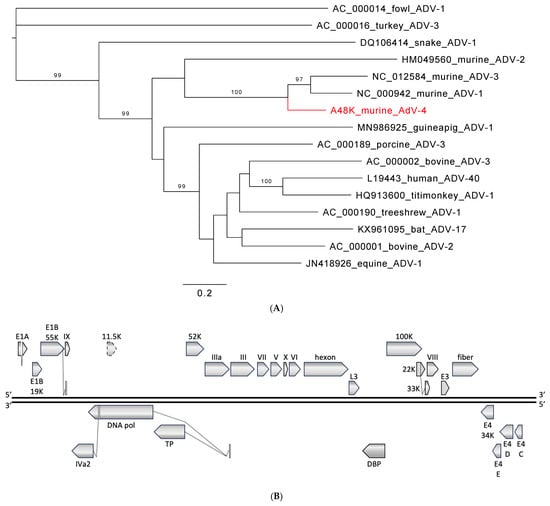
MAdV-1 and -2 have been isolated from laboratory mice and only MAdV-3 from a striped field mouse (Apodemus agrarius) [15,16]. Pathology and tissue tropism for the three viruses are mainly known from experimental infections. MAdV-1 is primarily related to central nervous system (CNS) infection, but the virus also spreads to the kidney, spleen, and lung. However, outcomes vary with mouse strain, animal age, and route of infection [77,78]. MAdV-2-infected animals commonly appear healthy, and the virus is found in the intestinal tract and not in the lung, liver, or urine [15,16,79,80]. MAdV-3 was isolated from liver tissue, although the highest load was found in the lung of the striped field mouse [15]. It has been hypothesized for MAdV-1 that its residual pathogenicity might reflect an incomplete adaptation to the mouse host and a recent switch from an alternative host, while MAdV-2 is thought to be a genuine virus of mice that coevolved with its host. Little is known about murine adenoviruses in wild rodents. Small sequence fragments (~300 bp) obtained by nested PCR from feces have been reported from wild rodents in China (MAdV-2 [81] and MAdV-2, -1, -3 [31]) and by approaches in the USA (MAdV-2 [38]), and only two studies provide longer sequences (MAdV-1 [36] and MAdV-2 [25]). Similarly, MAdV-2-related, PCR-amplified sequences were reported from the dried blood spots of four rodents (Cricetomys (2), Hybomys, and Praomys spp.) in the Democratic Republic of the Congo, from the lung tissue of two multimammate mice in Kenya, and from the tissues of four rats in Côte d‘Ivoire [17,18,19]. In our study, we identified a novel fourth murine adenovirus in wild rodents, MAdV-4. Zoonotic transmission of MAdV-4 does not appear highly likely given the supposed species-specificity of adenoviruses and the sequence divergence from human mastadenoviruses [14]. However, there are reports that show or suggest transmission between human and non-human primates [82,83] and to and among animal species [12,19,84,85]. Recombination is an evolutionary mechanism in adenoviruses that may lead to sudden changes in host range, and findings of human adenoviruses in domestic animals support this notion [37,86,87]. However, recombination is not readily detected by common diagnostic assays that target one or a few genes of the virus, emphasizing the need for non-targeted, whole-genome detection approaches.
The genus Chaphamaparvovirus is composed of a rapidly growing group of related parvoviruses that have been identified in highly divergent species, including vertebrates and invertebrates. The type species Rodent Chaphamaparvovirus1 includes two viruses, MuCPV from house mice trapped in New York City for which the disease state was not assessed [25] and MKPV, which was shown to cause inclusion body nephropathy (IBN) in laboratory mouse strains presumably worldwide [22]. The latter study also provided evidence that transcriptional activity and pathology are restricted to the kidney, although MKPV sequences can be found in other tissues like the liver or lung (possibly related to latent infection) [21,22,23,24]. Related viruses have been retrieved from kidney samples of vampire bats (Desmodus rotundus; Desmodus rotundus chapparvovirus, DrChPV) [22,88] and a capuchin monkey (Cebus capucinus imitator; capuchin kidney parvovirus, CKPV) [22], compatible with a more generalized nephro-tropism for these viruses. MKPV has been shown to express in the kidney a p10 protein at high levels from the most 5′ ORF of the genome. Conserved p10 ORFs are present in MuCPV, TdChPV2, and the virus a non-human primate, CKPV, but not in kidney-derived DrChPV from bats that map in an adjacent clade (see Figure 2). Given the relationship between these viruses and the diversity of hosts in which they have been found, it remains unclear to what extent these represent true virus reservoirs or incidental infection of susceptible hosts. As such, virus naming by species of encounter may be misleading. Both viruses from Zambia, Mwangazi virus and Nyamadzi virus, were present in kidney samples (Mwangazi virus was also in the lung), and both have a p10 ORF and map in a monophyletic clade together with MKPV, TdChPV2, CKPV, and UaPV (from the kidney of a black bear). For UaPV, only a 5′-truncated sequence is available that does not allow the identification of the (presumably present) p10 ORF [89]. Given the relationship between these viruses, phylogenetically and through a common p10 ORF, inter-species transmission appears conceivable and, in view of the likely nephro-tropic infection even in a non-human primate, of sufficient concern to include them in surveillance efforts.
Limitations of this study include the limited sample size and the lack of matching excreta. However, our study of Zambian rodents identified viruses from several diverse viral taxa that are, for various reasons, of potential zoonotic relevance. The applied VirCapSeq-VERT approach is a positive enrichment augmentation to next-generation sequencing that targets all viruses known to infect vertebrate hosts and thereby results in approximately 1000-fold increased sensitivity [39,40]. As shown, it is sample type- and pathogen-agnostic and capable of identifying partial or complete viral genomes, as well as novel sequences highly divergent from known sequences. It currently represents the most suitable approach to detect any virus in low copy number or present in poorly preserved samples, and thus presents a powerful tool for comprehensive pre-pandemic viral surveillance.
Source link
Lavel C. Moonga www.mdpi.com