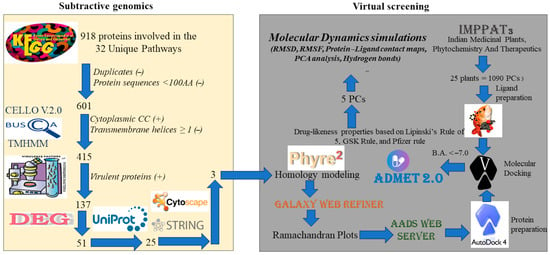
The subtractive genomics approach identified three potential drug targets that were exploited to identify chemical entities from a library of bioactive phytochemicals that can interfere with protein functions and potentially reduce the bacterial load of S. sputigena in periodontitis, providing potential novel therapeutics to treat this disorder. Accordingly, a series of computer-based techniques were integrated to screen the chemical library described above.
3.2.2. Phytochemical Screening
After the identification of the potential drug targets of
S. sputigena, we conducted a virtual screening of a natural product database to identify the most promising compounds that could potentially interact with the selected drug targets. In this chapter, we discuss the most promising hit compounds selected based on the binding affinity and the drug-like profile. The results of the screening are reported in
Table S1. The phytochemical (4bS,8aR)-2,4b,8,8-tetramethyl-7,10-dioxo-5,6,8a,9-tetrahydrophenanthrene-3-carboxylic acid (IMPHY005303) showed an interesting pattern of interaction, establishing polar contacts within the selected binding site of the protein typified by the code C9LUR0. In particular, IMPHY005303 formed four H-bonds with residues Asn48, Ser52, and Phe51 (
Figure 5A). This binding mode accounted for a calculated binding affinity of −7.2 kcal/mol (
Table 3).
Considering the other phytochemicals, pluviatilol (IMPHY006624) was able to interact with the binding site of the protein typified by the code C9LUR0, showing a docking score of −7.0 kcal/mol, which was slightly higher than that of the previously discussed compound (
Table 3). In particular, the compound could target residues Asn48, Arg59, Tyr138, Ala166, and Lys167 via H-bonds. In addition, we observed further contacts indicated as carbon–H-bonds with residue Ser165. Hydrophobic interactions with Tyr138 (π–π stacking interaction and π–alkyl contact) were detected. Alkyl interactions with Ala166 and Lys167 stabilized the retrieved binding mode (
Figure 5B). The interaction colored in red between the OH group and the Gln46 residue in the image indicates an unfavorable interaction, likely due to steric hindrance or unfavorable electrostatic interactions.
The next compound, 5-[3-(1,3-Benzodioxol-5-yl)-1,3,3a,4,6,6a-hexahydrofuro [3,4-c]furan-6-yl]-1,3-benzodioxole (IMPHY014895), showed a docking score of −7.6 kcal/mol within the C9LUR0 binding site (
Table 3). Polar interactions were detected with residues Ala53, Glu54, Tyr138, Lys167 (H-bonds), and Leu58 (carbon–H-bond). Various hydrophobic contacts were detected with Ala53, Thr57, Leu58, Arg59, Tyr138, and Lys167 (
Figure 5C).
Figure 5D shows the computational assessment of gadain (IMPHY004244) within the C9LUR0 binding site. The compound formed H-bonds with Ser52 and Arg59, whereas hydrophobic interactions were found with residues Arg59 (π–sigma and π–alkyl) and Tyr138 (π–alkyl). This binding mode resulted in a docking score of −7.1 kcal/mol (
Table 3).
The molecular docking output of the last compound (cubebin, IMPHY001912) is shown in
Figure 5E. This compound was able to establish polar and hydrophobic contacts within the selected binding site of the protein typified with the code C9LUR0. A canonical H-bond was detected with Ser52, whereas a carbon–H-bond was evident with residue Phe51. A strong network of hydrophobic interactions was established by IMPHY001912 with Arg59 and Tyr138. The docking score for this binding mode was −7.1 kcal/mol (
Table 3). The 3D models of molecular docking output regarding the protein typified with the code C9LUR0 are provided in
Figure S10.
Compound IMPHY005303 showed a docking score of −8.6 kcal/mol when docked into the binding site of the protein C9LRH1 (
Table 3). In particular, the compound was able to establish H-bonds with residues Thr253, Leu206, and Arg208 (
Figure 6A). Further hydrophobic interactions were detected with residues Leu132, Val205, Tyr209, and Leu232.
IMPHY006624 within the C9LRH1 binding site established a H-bond with Arg171 and carbon–H-bonds with Asp134 and Leu206. Hydrophobic interactions with Tyr172 (π–alkyl), and further contacts with residues Leu132 and Asp134 were detected (
Figure 6B). This binding mode resulted in a docking score of −8.3 kcal/mol (
Table 3).
IMPHY014895 established polar and non-polar contacts within the C9LRH1 binding site. In particular, it established a H-bond with Lys30 and a carbon–H-bond with residue Gly258. Different hydrophobic contacts were detected using Val205. Furthermore, additional electrostatic interactions were observed with residues Arg20 and Asp134 (
Figure 6C). The docking score value of −8.7 kcal/mol was related to the described binding mode (
Table 3).
IMPHY004244 exhibited a docking score of −8.6 kcal/mol within the C9LRH1 binding site (
Table 3). IMPHY004244 established several hydrophobic interactions: π–π stacking with Tyr172 and Tyr209, two alkyl interactions with Leu132 and Leu232 residues. Four π–alkyl contacts with Tyr172, Val205, Tyr209, and Leu232 (
Figure 6D).
Finally, compound IMPHY001912 was found to be the best performing compound considering the C9LRH1 enzyme. IMPHY001912 showed a docking score of −9.0 kcal/mol (
Table 3). Several contacts were found between the selected binding site and IMPHY001912. Two H-bonds and a carbon–H-bond were observed with Glu192, Thr233, and Asp135, respectively. Numerous hydrophobic contacts were established from IMPHY001912 with the following residues: Tyr172 and Tyr209 (π–π stacking and π–alkyl contacts), Leu132 (π–alkyl and alkyl contacts), Leu232, and Ile236 (alkyl contacts) (
Figure 6E). The 3D models of molecular docking output regarding the protein typified with the code C9LRH1 are provided in
Figure S11.
The last examined target was the protein typified by the code C9LTU7. The compound IMPHY005303 showed a docking score of −8.3 kcal/mol within the binding site of the protein (
Table 3). H-bonds were observed among the compound and residues Ile83, Asn103, Thr104, and Ala105. A π-donor–H-bond was established with Gly82, and a π–alkyl hydrophobic contact with His56 was detected (
Figure 7A). The interaction colored in red between the OH group and the Gly81 residue in the image indicates an unfavorable interaction, likely due to steric hindrance or unfavorable electrostatic interactions.
Figure 7B illustrates the docking output of IMPHY006624 within the binding site of C9LTU7. H-bonds were observed with residues Asp130 and Ala205, and a carbon–H-bond was evident with Ala205. IMPHY006624 was further able to establish two π–anion electrostatic interactions with residues Asp130 and Asp177. Furthermore, three alkyl hydrophobic bonds were formed with Leu50, Ala130, and Leu170, and a π–alkyl bond with Ala226 (
Figure 7B). This binding mode accounted for a docking score of −8.0 kcal/mol (
Table 3).
IMPHY014895 showed a docking score of −9.0 kcal/mol with the C9LTU7 enzyme (
Table 3). H-bonds were observed with residue Ser202, and a carbon–H-bond was evident with Asp130. A π-donor–H-bond was established with Gly82. Moreover, a π–anion electrostatic bond with Asp130 was observed. Further hydrophobic contacts were found with residues Leu50 and Ala105 (
Figure 7C).
Next, the compound IMPHY004244 established a carbon–H-bond with Thr172, a π–anion electrostatic bond with Asp130. Three alkyl bonds with residues Leu50, Arg84, and Ala128. Further π–alkyl bonds with Leu50, His56, and Ala128 were observed (
Figure 7D). The described binding mode accounted for a docking score of −8.2 kcal/mol (
Table 3).
Finally,
Figure 7E shows the docking output of IMPHY001912. Considering this target, the mentioned compound achieved the best computational score in terms of the prediction of affinity within C9LTU7 (
Table 3). In fact, a docking score of −9.1 kcal/mol was related to the following binding mode. IMPHY001912 established H-bonds with Asp11 and Ala205. Two π–anion electrostatic interactions were observed with residues Asp130 and Asp177. Furthermore, several hydrophobic contacts were detected with residues Leu50, Ala128, Ala205, and Ala226, indicating a significant predicted affinity of IMPHY001912 for the selected target. The 3D models of molecular docking output regarding the protein typified with the code C9LRH1 are provided in
Figure S12.
3.2.4. Molecular Dynamic Simulations
To provide a comprehensive overview of the binding mode of IMPHY001912, which was found to be the most promising ligand for the selected proteins, we conducted MD simulation studies. Apart from visually examining the generated trajectories, the trajectories were evaluated by computing the RMSD and RMSF values and conducting analysis of the dynamic ligand interaction graphs. Overall, the MD simulation considering the complexes C9LRH1/IMPHY001912 and C9LTU7/IMPHY001912 showed satisfactory stability of the binding mode of IMPHY001912 within the mentioned proteins, exhibiting small RMSD values and slight variations in the proteins, as shown by the RMSF. In contrast, when the complex C9LUR0/IMPHY001912 was examined, MD simulation analysis provided a relevant uncertainty concerning the binding mode of IMPHY001912, with limited stability and high RMSD, indicating a very weak possibility of interaction with the selected protein. Accordingly, IMPHY001912 may function as an inhibitor of the proteins C9LRH1 and C9LTU7, but not for the protein C9LUR0, based on the stability of the biological systems and the analysis of the pattern of interaction within the chosen binding sites throughout the course of the 100 ns MD simulations.
A detailed analysis of complex C9LRH1/IMPHY001912 is presented in
Figure 8 and in
Figure S13A. In particular, IMPHY001912 established a strong hydrophobic contact network with Val130, Leu132, Leu190, Val205, Thr209, Leu232, and Ile236. Furthermore, significant polar contacts, mainly H-bonds, with the side chains of Asp135 and Thr233 and with the backbone of Gly173 were observed. Overall, the MD simulation output supported the docking results, indicating significant interactions within the selected binding site, suggesting the potential of IMPHY001912 to inhibit the protein typified with the code C9LRH1. The region with higher RMSF (
Figure 8B) comprises approximately twenty residues structured in a loop that is not close to the binding site.
The MD simulation output for the complex C9LTU7/IMPHY001912 is shown in
Figure 9 and in
Figure S13B. Hydrophobic contacts with Val48, Leu50, Ala128, Leu170, and Leu224 were particularly evident during the simulation. In addition, a network of polar contacts (H-bonds) with Cys9 and Asp11 and an ionic bond with Asp130 were observed. Accordingly, the MD simulation output confirmed the docking results, indicating relevant interactions within the selected binding site, suggesting the potential of IMPHY001912 to act as an inhibitor of the protein typified with the code C9LTU7.
The MD simulation output for the complex C9LUR0/IMPHY001912 is shown in
Figure 10 and
Figure S13C. In contrast to the two previously discussed complexes, IMPHY001912 appeared to have not a significant affinity for this selected protein. In particular, as indicated by the graphs regarding the MD trajectory, the ligand showed high instability and was not able to reach a stable bioactive conformation; instead, several conformational changes were observed, precluding strong binding with the selected binding site (no contacts that occurred more than 30.0% of the simulation time were observed). Furthermore, after approximately half of the simulation time, IMPHY001912 lacked the capacity to interact with the selected binding site and disappeared from the selected binding site without any contact within the protein. These results were also obtained by considering two additional independent MD runs. Accordingly, IMPHY001912 showed limited affinity for the protein typified by the code C9LUR0, suggesting that it could not inhibit the aforementioned protein.
To further corroborate the MD simulation results, we determined the free binding energy (ΔG
bind) considering the entire MD trajectory of the selected complexes, providing a comprehensive picture of the binding affinity. Accordingly, by employing the MM/GBSA techniques, we obtained the ΔG
bind values listed in
Table 5. The main energy contributions to the overall binding free energy, taking into account various energy sources, are included in
Table 5. The van der Waals energy contribution was found to be the main source of the ligand binding energy considering the targets C9LRH1 and C9LTU7. This result emphasizes the critical role that hydrophobic interactions play in the stability of ligand/protein complexes, given the hydrophobic nature of the binding sites. On the contrary, as expected by analyzing the trajectory of the complex C9LUR0/IMPHY001912, we observed a dramatic decrease in the affinity for the compound IMPHY001912, indicating a very weak affinity for the considered target. In summary, based on the MD analysis and the calculated ΔG
bind, we conclude that IMPHY001912 (cubebin) could have a multitarget behavior because of the strong binding for the targets C9LRH1 and C9LTU7, whereas the affinity for the target C9LUR0 was limited.