1. Introduction
Fumonisins are mycotoxins primarily produced by molds of the
Fusarium genus, with
F. verticillioides (formerly
F. moniliform) being the most prevalent producer. These toxins are found globally, mainly in corn and corn-derived products [
1,
2]. The B subfamily contains the most abundant and toxic fumonisins, with fumonisin B1 (FB1) being the most prevalent. The toxicological effects of fumonisins vary widely among species. In horses, they cause neurotoxicity, leading to equine leukoencephalomalacia [
3]. In pigs, the lungs are the primary target, resulting in porcine pulmonary edema [
4]. In most animal species, fumonisins induce liver or kidney toxicity, with liver damage being particularly pronounced in poultry and mice, while nephrotoxicity is more common in rats. Fumonisins are known to be hepatocarcinogenic in mice and nephrocarcinogenic in rats, and they are classified by the International Agency for Research on Cancer (IARC) as group 2B—possibly carcinogenic to humans [
1,
5]. A provisional maximum tolerable daily intake (PMTDI) for fumonisins B has been established for humans, and guidelines for maximum levels in animal feed have been set [
1,
6].
Fumonisins are known to induce a variety of cytotoxic effects, with the disruption of sphingolipid synthesis being the most extensively studied [
7]. This effect stems from the structural similarity between FB1 and sphingoid bases, leading to numerous cellular alterations across multiple species. A hallmark of these changes is the significant variation in the sphingolipid profile observed in poultry [
8]. While the primary effects of fumonisins on ceramide synthases (CerS) can explain many of the cellular changes [
7], additional mechanisms of toxicity have been proposed [
2,
9,
10,
11,
12]. These include altered apoptosis, changes in cell proliferation, oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction, and disruptions in membrane integrity [
13]. Although it is challenging to discern whether these alterations are a direct cause or a consequence of fumonisin toxicity, it is clear that disturbances in lipid homeostasis are a consistent feature of fumonisin-induced cellular damage. Beyond sphingolipids, fumonisins also affect the levels of cholesterol, triglycerides, inositol phosphates, and polyunsaturated fatty acids (PUFAs) [
13]. Paradoxically, no study to date has comprehensively examined the effects of fumonisins on the oxylipidome.
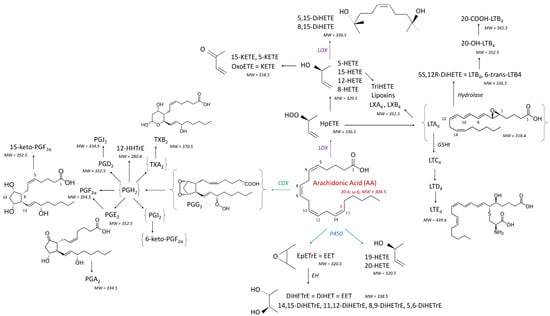
Oxylipins (OLs) are bioactive lipids formed through the oxidation of PUFAs and play critical roles in various biological processes, with their functions varying depending on the specific PUFA precursor [
14]. OLs derived from ω6-PUFAs, such as arachidonic acid (AA), linoleic acid (LA), gamma-linoleic acid (GLA), and dihomo-gamma-linoleic acid (DGLA) (
Figure 1 and
Figure 2), often have pro- or anti-inflammatory effects. In contrast, OLs produced from ω3-PUFAs like alpha-linoleic acid (ALA), eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA), and docosahexaenoic acid (DHA) (
Figure 2) are predominantly considered anti-inflammatory or neutral in nature [
15,
16].
The biological activity of OLs is also influenced by the metabolic pathway through which they are synthesized. OLs formed via the cyclooxygenase (COX) pathway from AA are mainly prostaglandins (PGs), which are typically pro-inflammatory. In contrast, OLs produced through the lipoxygenase (LOX) pathway can possess both pro- or anti-inflammatory properties (
Figure 1). For example, 15-HpETE, a precursor to 15-HETE, exhibits both pro- and anti-inflammatory roles, while also serving as a precursor to lipoxins (
Figure 2), which are specialized mediators involved in the resolution of inflammation [
17]. Similarly, OLs derived from cytochromes P450 (P450) display diverse roles in inflammation. For instance, 20-HETE is pro-inflammatory, while EpETrE is anti-inflammatory. Their metabolites, DiHETrE, formed through the action of epoxide hydrolase (
Figure 1), are pro-inflammatory, and the DiHETrE:EpETRE ratio increases during inflammatory responses [
18,
19,
20,
21]. Interestingly, while COX-mediated OLs from EPA are generally low in pro-inflammatory activity [
14,
22], the majority of OLs derived from EPA, DPA, and DHA through the LOX pathway are resolvins, protectins, and maresins (
Figure 2), which are specialized mediators involved in the resolution of inflammation [
17]. OLs can also be formed through the non-enzymatic (NE) oxidation of PUFAs, which typically occurs during oxidative stress within cells [
23]. As a result, a variety of HPLC-MSMS techniques have been developed to assess OL variations in living organisms. These methods provide insights into OL content specific to particular PUFAs, metabolic pathways, or inflammatory roles [
24,
25,
26,
27,
28].
The diverse effects of fumonisins, particularly their ability to reduce PUFA concentrations in cells [
13], suggest that these toxins may disrupt the oxylipidome. Since a wide array of OLs with distinct biological functions are generated from various PUFAs within cells [
14], a thorough examination of the oxylipidome—focusing on OLs derived from different PUFAs via multiple metabolic pathways—is highly warranted. To investigate this, we utilized an HPLC-MSMS method to profile 111 OLs, including 15 deuterated standards, originating from seven PUFAs through the COX, LOX, P450, and NE pathways (
Figure 1 and
Figure 2). Chickens were fed a diet containing 20.8 mg FB1 + FB2/kg for four and nine days with OL levels measured in both the liver and brain. This dose was chosen because it corresponds to the maximum of 20 mg FB1 + FB2 recommended by the EU in poultry feed [
29]. An exposure period of 4 to 9 days was chosen to reveal the early effects of fumonisins. The liver was selected for analysis due to the accumulation of FB1 over time in this organ [
30], which is the most sensitive to fumonisin-induced alterations in the sphingolipidome in chickens [
8,
31]. The brain was also studied, even though FB1 is not detectable in this organ, because fumonisins have been shown to alter the sphingolipidome in the brain in ways that cannot be fully explained by CerS inhibition alone [
32].
2. Materials and Methods
2.1. Reagents and Chemicals
All reagents and analytes were sourced from Sharlab (Sharlab S.L., Sentmenat, Spain) or Sigma (Sigma-Aldrich Chimie SARL, Saint Quentin Fallavier, France). Solvents for oxylipin separation via HPLC-MSMS were of LC-MS grade, while all other reagents and solvents were HPLC grade. Indomethacin, triphenylphosphine, butylated hydroxytoluene, and AUDA were purchased from Sigma. OLs used as standards or internal standards (IS) were supplied by Interchim (Interchim, Montluçon, France) and correspond to Cayman chemical products (Cayman Chemical, Ann Arbor, MI, USA). The IS mixtures included the Primary COX and LOX MaxSpec
® LC-MS Mixture, containing 6-keto-prostaglandin F1α-d4, prostaglandin F2α-d4, prostaglandin E2-d4, prostaglandin D2-d4, thromboxane B2-d4, 15(S)-HETE-d8, 12(S)-HETE-d8, and (d8)5-HETE; the Deuterated Arachidonic Acid CYP450 Metabolite MaxSpec
® LC-MS Mixture, containing 20-HETE-d6, (±)14(15)-EET-d11, and (±)11(12)-EET-d11 each at a concentration of 1 µg/mL in ethanol; and the Deuterated Linoleic Acid Oxylipins MaxSpec
® LC-MS Mixture, containing (±)12(13)-DiHOME-d4, 13(S)-HODE-d4, 13-OxoODE-d3, and (±)12(13)-EpOME-d4 each at a concentration of 10 µg/mL in ethanol. Additional OL standards, as detailed in
Table S1, were also sourced from the same supplier.
2.2. Fumonisin Diets and Sampling Protocol
The control diet, free from mycotoxins, and the fumonisin-containing diet were formulated using soya, wheat, and corn to meet the nutritional needs of the animals [
30]. The fumonisin diet was prepared by incorporating maize naturally contaminated with fumonisins, achieving final concentrations of FB1, FB2, and FB3 of 15.2, 5.6, and 0.9 mg/kg, respectively. The mycotoxin levels in raw materials and diets were quantified by LC-MSMS according to the AFNOR standard V03-110 [
33]. Among the 44 mycotoxins tested, only fumonisins were detected at significant levels [
30].
This animal study was conducted at Cebiphar (Cebiphar, Fondettes, France), as detailed in [
30], under project number V9152, project 2017062111426641, approved by the French Ministry of Higher Education, Research and Innovation on 6 November 2017. A total of 30 Ross broilers were randomly assigned into three groups of 10: one group receiving the mycotoxin-free control diet (Con), and two groups receiving the fumonisin-containing diet for four days (FB_4d), from day 17 to day 21, and for nine days (FB_9d), from day 12 to day 21. On day 21, the fumonisin diet was withdrawn eight hours prior to the sacrifice of the animals, and tissue samples were collected. Samples were stored at −80 °C until further analysis. No effects on performance or biochemical parameters were observed, as previously reported [
30].
2.3. Preparation of Standards, Chromatographic Separation, and Mass Spectrometry
Stock solutions of OLs were prepared at a concentration of 10 µg/mL in ethanol and stored at −20 °C or −80 °C, following the manufacturer’s recommendations. Calibration solutions were prepared with concentrations of 1000, 500, 250, 125, 62.5, 31.25, 15.63, 7.81, 3.91, 1.95, 0.98, 0.49, 0.24, and 0.12 ng/mL in ethanol for all analytes, except for 13-HODE and 9-KODE, which were also prepared in concentrations of 20, 10, 5, 2.5, and 1.25 µg/mL in ethanol. These calibration solutions were freshly prepared before each use. A mixture of deuterated standards was also prepared (v/v/v) to serve as internal standards (ISs).
Chromatographic separation was performed using Poroshell 120 column (3.0 × 50 mm, 2.7 μm). Analytes were eluted using two mobile phases: (i) water/acetic acid (100:0.1,
v/
v, phase A); (ii) acetonitrile/isopropanol (90:10,
v/
v, phase B). Separation was conducted at a flow rate of 0.3 mL/min for 30 min at 40 °C with the following gradients: 0–3.5 min from 15% B to 33% B, 3.5–5 min to 38% B, 5–7 min to 42% B, 7–9 min to 48% B, 9–15 min to 65% B, 15–17 min to 75% B, 17–18.5 min to 85% B, 18.5–19.5 min to 95% B, 19.5–21 min to 15% B, and 21–30 min to 15% B [
27]. Mass spectrometric detection was carried out using negative electrospray ionization under the following conditions: temperature 300 °C, flow rate of 10 L/min, pressure 25 psi, and capillary voltage 4000 V. Transitions, fragmentor voltages, and collision energies were optimized for each analyte using the Agilent MassHunter Optimizer software 2011 G3335-60091, with the results reported along with the retention times in
Table S1. Data analysis was performed using Agilent MassHunter quantitative analysis software B.05.291.0. The accuracy of the method was considered acceptable, with a relative standard deviation (RSD) of less than 20%.
2.4. Stability, Linearity, and Limit of Quantitation
The stability of the standard solutions of OLs in ethanol was assessed at room temperature over periods of 8, 16, 24, and 32 h, with concentrations of 0.5, 2, 8, 31, 125, and 500 ng/mL (
Table S2). Except for 5,6-EpETrE, which was deemed unstable, all other analytes remained stable for more than 24 h. The stability of 5-HpETE was 88% at 8 h and 65% at 24 h.
The linearity of the method was evaluated using OL solutions in ethanol at concentrations ranging from 0.1 to 1000 ng/mL, corresponding to 1 to 10,000 pg on column (
Table S3). For 13-HODE and 9-KODE, to account for the high concentrations observed in the liver samples, the method was extended to concentrations of 10 and 20 µg/mL, respectively. Different concentration ranges were employed to address the variation in OL levels between the liver and brain (
Table S3). The results demonstrated excellent linearity across all the OLs analyzed in this study, consistent with the previous findings [
24,
25,
27,
28]. The lowest concentration tested, which provided an accuracy of 80–120%, was determined to be the optimal limit of quantitation (LOQ).
2.5. Extraction and Purification of Samples
OLs in liver and brain tissues were determined using tissue homogenates prepared at 4 °C. For homogenization, 1 g of liver or brain tissue was combined with 3 mL of phosphate buffer (0.1 M, pH 7.4) and processed in a Teflon-coated Potter glass. The homogenate was then centrifuged at 3000× g for 15 min at 4 °C, and the supernatant (S3000) was collected and stored at −80 °C until analysis.
Liver and brain S3000 homogenates were extracted using 60 mg Oasis HLB 3cc extraction cartridges. Prior to extraction, the homogenates were diluted in NaCl buffer and mixed with an antioxidant cocktail following the methodology outlined in [
24,
25,
27,
28] with minor modifications. Specifically, 1850–1800 µL of 0.9% NaCl was added to 50–100 µL of the liver or brain S3000 homogenate. Then, 10 µL of the antioxidant cocktail containing EDTA (2 mg/mL), indomethacin (2 mg/mL), BHT (0.2 mg/mL), and triphenylphosphine (0.2 mg/mL) in a water/methanol/ethanol solution (2:1:1,
v/
v/
v) was added, followed by 10 µL of AUDA (5 mg/mL in DMSO), 50 µL of 50% acetic acid, 170 µL of ethanol, and 30 µL of the IS mixture.
The mixture containing the OLs was passed over the columns under a maximum vacuum of 20 mm Hg. Following this, the columns were washed with 2 mL of 5% methanol and dried for 30 min. The OLs were subsequently eluted with 1 mL of methanol and 2 mL of ethyl acetate into a borosilicate glass tube containing 5 µL of glycerol/methanol (30:70, v/v). The eluate was dried at 40 °C, reconstituted in 200 µL of ethanol, centrifuged at 3000× g for 15 min, and filtered through 0.45 µm nylon membrane syringe filters before injection into the HPLC system.
Samples were processed in batches of twelve, each containing three control samples, three samples from chickens fed the fumonisins diet for four days, and three samples from chickens fed the fumonisins diet for nine days. The samples were injected alternately with a 10 µL volume, ensuring a maximum delay of 10 h between the thawing of tissue homogenates and the last injection into the HPLC system.
2.6. Recovery of Oxylipins in PBS
The recovery of OLs was assessed by spiking analytes into a solution of phosphate-buffered saline (PBS) and passing the spiked solution through the HLB columns, as described in the extraction and purification section. The concentrations tested ranged from 0.2 to 25 ng/mL PBS (0.1 M, pH 7.4), corresponding to 20 to 2500 pg spiked before extraction.
An acceptable recovery of 70 to 120%, accompanied by an RSD of less than 20%, was obtained for the majority of analytes tested (
Table S4). A recovery of 60 to 70% was considered acceptable as long as the RSD was under 20%. Analytes with recoveries of less than 60% or greater than 130% across the 1–25 ng/mL concentration range and with an RSD of less than 20% were considered difficult to quantify. This included 11-trans-LTD4, 6-trans-LTB4, maresin 1, 14,15-DiHETE, 13-HODE, 9-HODE, and 16-HDoHE. These analytes were treated as providing comparative data between groups, but the quantities measured were regarded as estimates rather than precise values.
Any analyte with a mean RSD greater than 25% across the 1–25 ng/mL concentration range was deemed non-quantifiable in this study. This applied to 15-deoxy-δ12,14-PGD2, LTB4, and 5,6-EpETrE. Furthermore, 13-HpODE and 15-HpETE were not detected after passing through the HLB columns and were therefore considered non-quantifiable in this study.
2.7. Matrix Effect, Recovery, and Repeatability of Internal Standards in Liver and Brain
Signal suppression and enhancement (SSE) were evaluated for the IS by the method area at two concentrations in the liver and three concentrations in the brain (
Table S5). SSE values outside the 80–120% range indicated a matrix effect, which was considered in the final concentration calculation. SSE at a concentration of 0.4 µg/g for the deuterated OLs from AA and 4 µg/g for the deuterated OLs from LA were deemed acceptable for all the analytes used as IS based on an RSD below 20%.
The recovery of IS on the HLB columns was measured for analytes spiked in PBS solution, as well as in liver and brain homogenates, as described in the extraction and purification section. A recovery of 70 to 120% was obtained in all three matrices for the majority of the IS (
Table S6). The recovery of (d3)13-KODE in PBS, liver, and brain was 71%, 50%, and 57%, respectively. This recovery was consistent with the mean recovery of 13-KODE in PBS, which was 68% (
Table S4). A very low recovery of (d8)15-HETE was observed in both PBS and tissues. Since the mean recovery of 15-HETE in PBS was 78% (
Table S4), we decided not to use (d8)15-HETE as an IS. The concentrations of 15-HETE in tissues were corrected by the recovery measured for (d8)12-HETE.
Intra- and inter-day repeatability were assessed by the RSD of the recovery measured for the IS (
Table S7). A value below 20% was considered acceptable.
2.8. Statistical Analysis
Statistical analyses were performed using XLSTAT Biomed 2018 software (Addinsoft, Bordeaux, France). To compare oxylipin concentrations in the liver and brain of chickens fed the control diet (n = 10) and the diet containing fumonisins for four (n = 10) or nine days (n = 10), a one-way ANOVA was performed after verifying homogeneity of variance using Hartley’s test. Differences among groups were considered statistically significant at p < 0.05. In cases where a significant difference was detected, individual means were compared using Duncan’s multiple range test. If variance homogeneity was not met, data were logarithmically transformed and re-analyzed. When variance remained non-homogeneous, the Kruskal–Wallis test was used. Statistically different means were indicated by different letters for the same variable.
The global effects of fumonisins on the oxylipidome were also evaluated using partial least squares discriminant analysis (PLS-DA). A Q2 value greater than 0.5 was considered indicative of a good predictive capacity for the model. Oxylipins with projection values greater than 1.1 were regarded as significant contributors to explaining the clustering of chickens into different groups.
5. Conclusions
In conclusion, fumonisins, even at a dose deemed safe, have a profound impact on the hepatic and brain oxylipidomes in chickens. In the liver, the effects of fumonisins were not dependent on the ω3- or ω6-character of the PUFAs but rather on the chain length, with a more pronounced increase in OLs derived from C18-PUFAs compared to those from C20 or C22 PUFAs. After four days of fumonisins feeding, a specific increase in OLs formed by the P450 pathway was observed, while at nine days, OLs from P450, along with several from the COX and LOX pathways, were also elevated. The diol/epoxide ratios were strongly reduced at both four and nine days of fumonisins exposure. In the brain, OLs derived from ω3-PUFAs were more indicative of fumonisins’ effects than those of ω6-PUFAs, particularly after four days of feeding the contaminated diet. The most markedly increased OLs were those enzymatically formed by the LOX and COX pathways, with a smaller increase from the P450 pathway. A variable rise in the diol/epoxide ratios was observed at both four and nine days. Collectively, these results suggest that the effects of fumonisins on OLs in the liver and brain are both specific and distinct. The changes in OLs in the liver are likely due to P450-mediated alterations and do not appear to be related to an inflammatory process, while the changes in the brain are indicative of an inflammatory/anti-inflammatory response, potentially linked to the effects of fumonisins on specialized cells, such as macrophages or glial cells. Further research is necessary to confirm these hypotheses.