1. Introduction
Human complexity and variability have limited our understanding of the pathobiology of disease causality, onset, and progression [
1]. It is salutary to consider that each of us consists of approximately ~37 trillion cells, and approximately 50% are microbial constituents of the “human microbiome” [
2]. We consist of ~200 different cell types [
3], possess ~20,000–25,000 genes [
4], 100,000–2,000,000 proteins (including isoforms) [
5], and >3000 metabolites [
1]. Even an organ such as a lung is biologically complex. There are ~40–60 cell types, including epithelial, endothelial, fibroblast smooth muscle, nerve, and assorted immune cells [
6]. The adult human lung contains approximately 480 million alveoli to effect efficient oxygen and carbon dioxide gas exchange. They provide a large surface area of ~70 square meters (the size of a tennis court) for efficient gas exchange to meet the body’s oxygen demands [
7]. In contrast, newborns have only ~20–50 million alveoli in their lungs at birth [
8]. This limited understanding due to complexity has resulted in the lack of safe and efficacious therapeutic drugs for treating numerous specific disease conditions [
1]. One example of such a complex disease state with meager therapeutic options is bronchopulmonary dysplasia (BPD) [
9].
BPD is the most common pulmonary complication in premature infants [
10]. Roughly 30–50% of extremely premature neonates develop BPD [
11]. More than 15,000 new cases of BPD are diagnosed each year in the United States, with annual costs estimated to be more than USD 2.5 billion [
12]. The impact of BPD on parents and their families, medical services, and society is significant [
13]. Prematurity with surfactant deficiency and a limited number of alveoli, a condition called respiratory distress syndrome (RDS), is the initial causative event for BPD. With the increased trend of premature births and improved medical management, more and more extremely premature neonates (<29 weeks of gestation) are now surviving, which contributes to the unchanged annual BPD prevalence. Implementing clinically tested BPD treatment, such as intramuscular vitamin A injection [
14] and early postnatal caffeine treatment [
15], does not reduce the BPD rate, exemplifying the poorly understood pathobiological BPD processes.
BPD’s three major postnatal contributors are supplemental oxygen, mechanical ventilation, and infection. All these contributors cause excessive oxidative stress (OS). The poorly developed antioxidant capacity in premature neonates makes them susceptible to OS. Judicious oxygen use, early surfactant therapy, gentle ventilation strategy, aggressive nutritional support, early postnatal caffeine treatment, and vitamin A injection have been widely accepted as standard management for premature neonates. The unchanged or slightly increased prevalence of BPD indicates, however, room for improvement, and new therapeutic modalities should be explored. A common approach for developing new therapeutic interventions is identifying novel pathobiological processes in the targeted disease condition. For example, the role of endoplasmic reticulum (ER) stress in BPD has recently been reported by us [
16], and this review, in part, assesses the potential of targeting pathway/network constraints in ER stress to treat BPD effectively.
The ER is a subcellular organelle synthesizing surfactants [
17], proteins, lipids, and cholesterols [
18]. It intimately interacts with other organelles, essential for cell homeostasis. The ER is sensitive to OS, however, which can result in ER stress [
19]. ER stress has been implicated in several lung disorders, including hyperoxia (HOX)-induced lung injury [
20]. Our recent report was the first to show increased ER stress in human BPD lungs [
16]. We further identified that ER stress is interrelated with autophagy, apoptosis, sterile inflammation, and cellular senescence [
21]. All these associated pathologic changes can inhibit alveolar formation and reduce the lung growth potential. In animal studies, we then demonstrated that caffeine and chemical chaperones attenuated the severity of HOX-induced rat BPD by ameliorating ER stress. Later, we identified the crucial role of myeloperoxidase (MPO)-induced OS in BPD [
22]. We demonstrated decreased ER stress in BPD rat lungs by treating rat pups with the reversible MPO inhibitor N-acetyl-lysyltyrosylcysteine amide (KYC) with improved alveolar formation [
21]. This new observation raises the possibility of using MPO inhibitors in ER stress-mediated disorders.
Despite these findings, it is essential to recognize that ER stress is not typically the primary cause of disease onset in BPD or other major disease states. Rather, ER stress represents a significant downstream consequence of various initiating factors, such as OS and inflammation [
9]. This distinction between continuum and causality has considerable ramifications for therapeutic development. Specifically, it raises important questions about whether targeting ER stress directly is the most effective strategy or if addressing the upstream causes of ER stress might yield better therapeutic outcomes. This nuance will be explored in depth throughout this review as we examine the role of ER stress in BPD within the broader context of disease pathogenesis. In this review, we focus specifically on BPD. We will start by describing the structure and function of the ER, followed by how OS elicits ER stress and the role of ER stress in BPD. We will then discuss the therapeutic potential of chemical chaperones and MPO inhibitors in BPD treatment.
5. ER Stress in BPD
ER stress has been demonstrated in several different diseases, such as diabetes, cancer, neurodegenerative disorders, and some lung disorders, including HOX-induced lung injury [
20]. The idea that ER stress contributes to BPD stems from the study by Choo-Wing et al., wherein their group showed increased ER stress in HOX mouse pup lungs and alveolar simplification. Although the term integrated stress response was used, the changes can also be explained as ER stress [
129]. Lu et al. used premature rat pups (E21) exposed to 85% oxygen to show an increased expression of CHOP and GRP78, apoptosis, and decreased radial alveolar count [
130]. Later, our group showed that increased ER stress in HOX-exposed rat pup lungs can be successfully attenuated with early caffeine treatment [
116]. In another report, we further demonstrated an increased ER stress in human and rat BPD lungs [
16] (
Figure 3). These human and animal studies strongly suggest ER stress plays a contributing role in BPD.
The improved alveolar structure with decreased ER stress in the BPD rat lungs by caffeine may not, however, support the role of ER stress in BPD [
116]. The concern stems from the fact that caffeine has been reported to induce ER stress [
131], although contradictory results have also been reported [
132,
133]. An ER inducer and a chemical chaperone were added to our animal studies to provide more evidence. Tunicamycin is a mixture of three naturally occurring antibiotics. By inhibiting UDP-
N-acetylglucosamine-dolichol phosphate
N-acetylglucosamine-1-phosphate transferase, it blocks the first step of protein N-glycosylation in the ER [
134], resulting in ER stress. Rat pups that receive a single intraperitoneal injection of tunicamycin develop a BPD phenotype characterized by alveolar simplification, increased apoptosis, myeloid cell infiltration, decreased capillary count, and increased myeloperoxidase (MPO) expression in the lungs reminiscent of HOX-induce BPD rat lungs [
16].
TUDC is an FDA-approved hydrophilic bile-acid derivative used to treat cholestasis with a chemical chaperone activity [
135]. It has been used in human neonates for cholestatic liver dysfunction, usually after prolonged total parenteral nutrition [
114]. As a complementary study, TUDC was given to HOX-exposed and tunicamycin-treated rat pups. The TUDC treatment successfully reduced ER stress, MPO expression, and myeloid cell infiltration and improved alveolar formation in both HOX-exposed and tunicamycin-treated rat pups (
Figure 4). Together with the results from tunicamycin-treated lungs, it was concluded that ER stress does contribute to BPD [
16].
Since MPO has been demonstrated to play a crucial role in HOX-exposed rat BPD [
16], and hypochlorous acid (HOCl) generated by MPO is a potent reactive oxygen species, we investigated whether inhibiting MPO activity can reduce ER stress in the HOX rat BPD model. Our study used KYC as the MPO inhibitor. The results demonstrate an attenuation of ER stress (
Figure 5) [
16] and cellular senescence [
21] in BPD rat lungs. Interestingly, one of the studies also showed the preservation of AT2 in the BPD rat lungs by both TUDC and KYC [
21]. Since the AT2 is considered a residential progenitor cell in the lung after the saccular stage [
136], this finding may contribute to a better lung growth potential for the HOX-exposed rat pups. We did not attempt to demonstrate whether KYC has any chemical chaperone activity, but its system pharmacology effects can explain its ability to attenuate ER stress [
22].
5.1. ER Stress Impairs Angiogenesis
There are at least two mechanisms by which ER stress can impair angiogenesis. First, some growth factors and their corresponding receptors and transporters require proper handling in the ER to obtain full function. One protein crucial to angiogenesis is the vascular-endothelial-growth-factor receptor type 2 (VEGFR
2) [
41]. N-glycosylation of VEGFR
2 occurs in ER to elicit pro-angiogenic signaling [
137]. ER stress will thus impair angiogenesis. Second, mitochondrial activity is supported by intimate support from the ER. Since normal mitochondrial function is required for angiogenesis, ER stress can again disturb angiogenesis. Other mechanisms by which ER stress can inhibit angiogenesis or cause aberrant angiogenesis include secondary OS, apoptosis, sterile inflammation, and cellular senescence. As adequate angiogenesis is critical to alveolar formation, we can expect ER stress to contribute to BPD.
5.2. ER Stress and Autophagy
Both ER stress and autophagy are stress response pathways that maintain cellular homeostasis. Under stress, the two pathways crosstalk and mutually activate each other to maintain an orchestrated response [
138]. They also work together to limit the pro-inflammatory response [
139]. If, however, the stress is excessive or sustained, these two pathways will jointly kill the cells by stabilizing the apoptosis effector PERP [
140].
5.3. ER Stress and Cellular Senescence
Cellular senescence is a biological phenomenon first described by Hayflick and Moorhead six decades ago [
141]. The topic recently attracted much attention due to its relationship with the aging process [
142]. Cellular senescence is not the same as aging; it also appears during fetal development and controls organ patterning [
143]. Senescence is also required during wound healing [
144]. A higher level of senescence is detected in the lungs during the late saccular stage [
145,
146]. The timely removal of senescent cells by phagocytic cells is the key for the orchestrated organ development and wound healing. Any stress that leads to DNA damage can initiate cellular senescence. OS, a significant contributor to BPD, is one of the most common mechanisms to elicit senescent change. Strong evidence suggests that ER stress tightly interacts with cellular senescence, but their temporal relationship remains to be determined [
147].
Behaviors of senescent cells include arrest proliferation, metabolic dysfunction, releasing inflammatory mediators, hypermetabolism, and transforming neighboring cells into senescent cells through the paracrine effect [
148]. In the HOX rat BPD model, we showed a senescent change in multiple lung cell types, including AT2 and endothelial cells [
21] (
Figure 6). These findings undoubtedly indicate that cellular senescence contributes to BPD. TUDC and KYC treatments can decrease ER stress [
16], attenuate cellular senescence [
21], and improve alveolar formation in the HOX BPD rat lungs. These encouraging findings suggest that ER stress can be a therapeutic target for BPD treatment.
5.4. ER Stress in BPD Destructive Cycle
Based on our recent findings initiated by OS, we have built a description of the BPD destructive cycle (
Figure 7). OS induces sterile inflammation with myeloid cell infiltration and MPO upregulation in the neonatal lungs, which results in a self-perpetuated destruction cycle involving ER stress and cellular senescence [
9]. The ER stress fuels the destructive cycle by generating more OS from refolding proteins and electron transport chain uncoupling from mitochondrial dysfunction. The inflammation and nutrient-deprivation activity of senescent cells will only aggravate the cycle more. We hypothesize that this is the reason why therapy targeting only one BPD mechanism may not be enough to reduce the BPD prevalence.
5.5. Opportunity to Attenuate ER Stress for Treating BPD
Unlike cellular senescence, which does not increase at the lung saccular stage but persists after BPD rat pups have returned to room air [
21], ER stress occurs as early as the lung saccular stage by OS [
16]. The temporal relationship between ER stress and cellular senescence suggests that ER stress contributes mainly to BPD onset, while cellular senescence contributes to BPD progression. That is to say that the state of the disease should determine the BPD treatment. Chemical chaperones might be efficacious in the early BPD stage, when patients are still under oxygen treatment, while senotherapy might be appropriate during the later stage of BPD. Compared to chemical chaperones and senotherapeutics, KYC, as a systems pharmacology agent, provides therapeutic efficacy, expanding from the onset to the progression of BPD, which may be a better therapeutic agent for BPD treatment.
5.6. Potential Therapies for ER Stress
Chemical chaperones are the most commonly used compounds for treating ER stress. Still, some compounds attenuate ER stress without stabilizing unfolded proteins directly. 4-PB, TUDC, and guanabenz are the only three FDA-approved chemical chaperones, and pioglitazone is the only FDA-approved non-chemical chaperone ER stress suppressor that can be used in humans. However, they have not been approved for treating human proteostasis perturbation.
Animal and human evidence indicates ER stress contributes to a wide variety of diseases, including diabetes [
149], neurodegenerative disease (Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease) [
150], asthma [
151], pulmonary fibrosis [
80], cystic fibrosis [
152], α-1 antitrypsin deficiency [
153], chronic obstructive pulmonary disease [
154], et al. Reports show that chemical chaperones or pioglitazone treatments might benefit proteostasis perturbation diseases.
TUDC is efficacious in treating diabetes [
155], neurodegenerative disorders [
150], and pulmonary fibrosis [
156]. In mice, TUDC has been shown to prevent progression from prediabetes to diabetes [
157,
158], diabetic visual deficits [
159], and endothelial dysfunction [
160,
161]. TUDC has also been shown to attenuate amyloid deposition in mouse models of Alzheimer’s disease [
162]. 4-PB is also known to have benefits for diabetes [
163]. There are extensive investigations into using chemical chaperones to treat proteostasis perturbations caused by destabilizing missense mutations [
95]. We should remember that some compounds do not have the properties of chemical chaperones but can attenuate ER stress by different mechanisms [
110]. These compounds include crocin, proanthocyanidins and anthocyanin, cordycepin, pioglitazone, and caffeic acid phenethyl ester.
A search on ClincalTrials.gov found 333 registered trials of chemical chaperones or ER stress suppressors on diabetes (322 for pioglitazone, 5 for TUDC, 4 for 4-PB, and 1 for crocin), 7 registered trials for cystic fibrosis (3 for pioglitazone, 3 for 4-PB, and 1 for TUDC), 6 registered trials for asthma (5 for pioglitazone and 1 for TUDC), 5 registered trials for Alzheimer’s disease (3 for pioglitazone, 1 for TUDC, and 1 for 4-PB), 3 registered trials for pulmonary fibrosis (all for pioglitazone), and 1 registered trial for Parkinson’s disease (pioglitazone), but no clinical trial for α-1 antitrypsin deficiency, COPD, and BPD. We are not surprised that there are no registered trials in ClincalTrials.gov or reported clinical studies for BPD using chemical chaperones or ER stress suppressors, as the role of ER stress in BPD has only been suspected just recently.
6. Discussion
The extensive interaction with almost all subcellular organelles has made the ER critical in cellular homeostasis. Its multitasking ability also makes it highly susceptible to endogenous and exogenous stress. The most commonly described stress that disrupts ER function is OS. OS elicits ER stress (or UPR), the focus of this review, with a myriad of downstream responses. Excessive ER stress has been described to play a role in multiple lung disorders, including HOX-induced lung injury [
20]. Using rodent models, our group and a few others have demonstrated that ER stress plays a role in developing the HOX-induced BPD phenotype. We showed that early postnatal caffeine treatment attenuates ER stress in the HOX rat BPD lungs and alveolar simplification [
116]. Since conflicting effects of caffeine on ER stress have been reported in the literature, we started to ask the question about what the role of ER stress in BPD is. This review is based on a literature review and our research results. We hope the work can provide adequate information to interested researchers in related fields.
This review starts by describing why ER stress is considered in the pathophysiology of BPD. It is followed by a focused perspective of the structure of the ER and how several groups of specialized membrane-bound proteins maintain its complex structure. Those specialized proteins mainly stabilize the high curvature tubular form of the ER. The first section of the second part of this review describes the primary biological functions of the ER, including protein synthesis, lipid and cholesterol synthesis, calcium regulation, detoxification, and several inter-organelle communications. The third part briefly describes a particular type of autophagy, ER-phagy, that pertains to the ER.
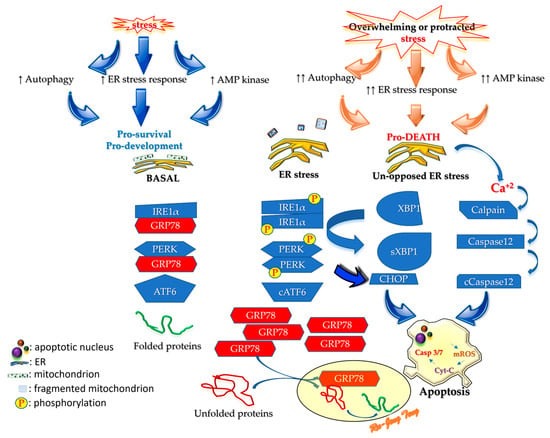
The third part of this review starts by summarizing ER stress and then describes pathways after it. The endogenous chaperone BiP/GRP78 is the most crucial modulator of ER stress. Under non-stressed conditions, BiP/GRP78 chaperones the ER stress sensors (IRE1α, PERK, and ATF6) to prevent dimerization, phosphorylation, and cleavage. In stressed conditions, the ER cannot handle protein folding, and this proteostasis perturbation forces BiP/GRP78 to leave the sensors to assist protein refolding. As protein refolding requires an oxidative environment, ER stress will generate OS that amplifies the stress. IRE1α and PERK start homodimerization and phosphorylation after freeing from BiP/GRP78 to activate the downstream cascades to halt the synthesis of non-essential proteins and speed up the translation of BiP/GRP78 to resume ER homeostasis. The liberated ATF6 will be cleaved by the Golgi apparatus into a transcription factor (
Figure 1) that prevents the protein synthesizing machinery from producing non-essential proteins. When proteostasis perturbation becomes unmanageable, excessive ER stress will initiate damaging processes, including inflammation, autophagy, apoptosis, ERAD, RIDD, and generating more OS.
The fourth part of the review is about chemical chaperones. Chemical chaperones are small molecules that can stabilize proteins non-specifically in specific harsh environments. Although some chemical chaperones have been studied in proteostasis disorders, no attempt has been made in the pediatric field. 4-PB, UDC, and TUDC are three chemical chaperones studied in children for diseases other than proteostasis disorders. Animal studies showed encouraging results in attenuating ER stress and BPD (
Figure 4). We believe chemical chaperones may have potential in BPD treatment.
The last part of this review starts by addressing evidence suggesting ER stress contributes to BPD. This evidence includes how ER stress impairs angiogenesis, eliciting autophagy and cellular senescence, which culminates in a BPD destructive cycle we proposed recently (
Figure 7) [
9]. The complexity of the stakeholder relationship explains why a single-agent therapeutic strategy does not work for BPD. The temporal relationship between ER stress and cellular senescence suggests their contribution to BPD differs at different disease stages. Our novel reversible MPO inhibitor—KYC—can attenuate ER stress and cellular senescence in BPD lungs. Besides the antioxidant and anti-antiapoptotic activities, KYC also preserves the AT2 cell count in BPD lungs. Our data strongly support KYC as a systems pharmacology agent [
128] with great potential in BPD treatment.
The considerations necessary for determining whether specifically targeting ER stress in BPD is a viable approach are numerous.
- (i)
If ER stress is a downstream event, targeting it may not resolve the underlying processes driving BPD, making it less valuable as a therapeutic target.
- (ii)
Causal vs. Consequential in Complex Pathways: Causal and consequential factors are not mutually exclusive in many diseases. Just because a process like ER stress arises as a consequence of other pathological events does not mean that addressing it would be ineffective. For example, even if inflammation and OS are the primary drivers of BPD, ER stress may still amplify the damage or serve as a critical mediator of cellular dysfunction. Therefore, therapies targeting ER stress might mitigate disease progression even if they do not fully address the initial cause.
- (iii)
Therapeutic Efficacy in Reducing Consequences: Even if ER stress is a consequence of BPD, it could still play a significant role in worsening disease severity by contributing to cell death, tissue damage, failure of repair processes, or even cellular senescence. In other diseases, such as neurodegenerative disorders, addressing downstream consequences (like protein misfolding or OS damage) has shown therapeutic value. Therefore, such an analysis is essential, since targeting a consequential process might still provide a therapeutic benefit by dampening a destructive feedback loop.
- (iv)
Multifactorial Disease Processes: BPD likely involves multiple interacting processes, like many complex diseases. Even if ER stress is not a primary driver, it may synergize with other pathological mechanisms, such as mitochondrial dysfunction or immune activation. Therapeutic approaches targeting multiple points in the disease cascade, including causes and consequences, often provide better outcomes than focusing on a single factor.
- (v)
The Importance of Timing: If ER stress is a consequence, the timing of intervention becomes essential. Early intervention targeting upstream processes might prevent ER stress altogether, whereas targeting ER stress later in the disease course could still alleviate symptoms or slow progression. Any analysis should factor in the stage of disease progression when evaluating whether targeting ER stress remains viable.
7. Conclusions
The frustratingly persistent prevalence of BPD has pressed us to seek new therapeutic strategies. Combining our literature search and our studies, we firmly believe ER stress contributes significantly to BPD. So many signaling pathways are involved in BPD that treatment focusing on one pathway will not be enough. The root causes of BPD are OS and inflammation [
97]. Unfortunately, premature neonates susceptible to BPD are born with increased OS and inflammation. Supplemental oxygen treatment and mechanical ventilatory support are life-saving for premature neonates. The ER stress results from OS and inflammation, which reciprocally augments OS and inflammation; we thus consider ER stress a reasonable therapeutic target for premature neonates. Although our data suggest that ER stress has an impact probably in the onset stage of BPD [
16], the employment of specific chemical chaperones should benefit neonatal lungs when premature neonates are still under oxygen support. The interactions between ER stress and other contributors suggest that chemical chaperones may still provide protection. Senotherapeutics may be helpful during BPD progression, as cellular senescence is not detectable at the saccular stage [
21]. KYC, which holds tremendous potential as a systems pharmacology agent, offers more protection than chemical chaperones or senotherapeutics. This novel tripeptide repurposes MPO into a catalase-like protein and upregulates NRF2-mediated antioxidative enzymes [
22] that attenuate ER stress [
16]. We coined the term ‘systems pharmacological agent’ for KYC due to its multi-layered protection against BPD. Transcriptomic studies show that KYC reverses key changes in hyperoxic BPD rat lungs, including increased leukocyte migration, chemotaxis, degranulation, and NETosis, while restoring WNT-catenin and Notch signaling (unpublished data). Although KYC seems promising in our animal studies, it has yet to be FDA-approved. Due to the size of the rat pups, the route of administration was intraperitoneal. Other routes of administration need to be investigated. Presently, KYC is under NIH-funded phase II SBIR animal studies for toxicology and routes of administration. We hope other systems pharmacology agents will soon be available for BPD treatment.