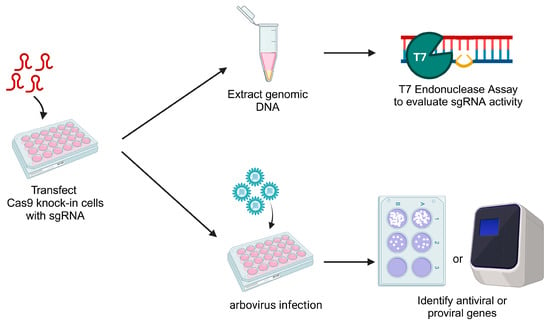
1. Introduction
Culex mosquitoes are vectors for a variety of pathogens that impact animal and public health, including West Nile virus (WNV) [
1], Saint Louis encephalitis virus [
2], and Usutu virus (USUV) [
3].
Culex mosquitoes are also vectors for the parasite
Plasmodium relictum, which causes avian malaria and has had significant impacts on Hawaii’s native bird populations [
4]. Currently, no vaccines are available for these pathogens [
5]. Despite their threat to public health, they remain understudied compared to
Aedes and
Anopheles mosquito species. Current vector control methods rely on the use of insecticides, but mosquitoes are becoming increasingly resistant to insecticides [
6]. New methods that utilize clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (Cas9) systems have been proposed and tested in several mosquito species to prevent disease transmission or reduce vector population [
7]. However, genetic-engineering-based strategies that affect disease transmission require an understanding of proviral and antiviral genes, and the molecular tools to assess gene function in
Culex species mosquitoes are limited [
7].
One avenue to reduce mosquito-borne pathogen transmission would be to manipulate mosquito immune genes via CRISPR/Cas9 editing. In mosquitoes, the antiviral defense is largely mediated by the RNA interference (RNAi) pathways [
8]. Dicer-2 (Dcr-2) is responsible for recognizing and cleaving viral double-stranded RNA (dsRNA) into small interfering RNAs (siRNAs), which are then loaded onto the RNA-induced silencing complex (RISC) [
9,
10]. Argonaute-2 (Ago-2) is a component of RISC and guides the degradation of viral RNA based on the siRNA sequences [
11,
12]. The loss of Dcr-2 in mosquitoes increases the replication of several arboviruses and insect-specific viruses [
13,
14,
15,
16,
17]. Similarly, disrupting Ago-2 expression increases virus replication [
17,
18,
19,
20] and may compromise the balance between mosquito immunity and virus replication. In
Culex quinquefasciatus-derived cells, another antiviral protein, Vago, is secreted following WNV infection and elicits an antiviral response [
21]. However, most of our understanding of antiviral responses in mosquitoes comes from studies performed in
Drosophila melanogaster flies or
Aedes aegypti mosquitoes. Less is known in
Culex species mosquitoes, and fewer molecular tools are available. RNAi has been used for decades to reduce gene expression in mosquitoes and cells, but it is not always very efficient and using an antiviral pathway to study antiviral responses can be confounding. One of our recent studies investigated the effects of silencing multiple
piwi genes in
Culex quinquefasciatus cells, where we observed a trend of
piwi5 silencing increasing Usutu virus (USUV) replication, but the effects were minimal and highly variable [
22]. Having more efficient methods to reduce gene expression may help to elucidate whether genes such as
piwi5 are, in fact, antiviral.
Recently, studies have validated the feasibility of CRISPR/Cas9 editing in the
Cx. quinquefasciatus genome, both in mosquitoes through embryo injection or REMOT [
23,
24,
25,
26], and in cell cultures through plasmid-based methods [
27,
28]. Briefly, CRISPR/Cas9 systems work via a single guide RNA (sgRNA) directing a Cas9 endonuclease to a complementary DNA target sequence, which Cas9 cleaves to generate a double-stranded DNA break. DNA repair mechanisms are error-prone and can result in insertions, deletions, or mutations that prevent gene expression [
29]. We previously optimized a plasmid construct for the transient expression of Cas9 and an sgRNA in
Cx. quinquefasciatus cells, which enabled the generation of gene knockout cells without constitutive Cas9 expression [
27]. In contrast, Feng et al. (2021) used site-directed homology to generate a
Cx. quinquefasciatus mosquito line expressing Cas9 in germline cells [
26]. This mosquito line was made via the injection of a single plasmid that contained Cas9 under the control of the germline-specific
vasa promoter, a dsRed fluorescent marker, and both flanked by homology regions to the
cardinal gene locus. Additionally, the plasmid contained a necessary sgRNA cassette for targeted insertion. The mosquito line was found to successfully edit the
kh gene when an sgRNA was introduced [
26].
While Cas9-expressing mosquito lines can be used to understand the genetic mechanisms important to vector competence and disease transmission and will be essential for developing transgenic mosquitoes, administering gRNAs to knockout-specific genes in mosquitos can be technically difficult. Additionally, the upkeep of multiple transgenic mosquito lines requires space and labor. Determining gene function in cell cultures before in vivo validation in mosquito lines can serve as a highly beneficial proof of concept. CRISPR/Cas9 plasmid-based methods have offered an avenue to study gene function in mosquito cell lines. CRISPR/Cas9-edited mosquito cells have been developed previously, including an
Aedes aegypti-derived Aag2 Dcr-2 knockout cell line, an Aag2 Argonaut 2 (Ago-2) knockout cell line, and an
Aedes albopictus-derived C6/36 Nix knockout cell line [
30,
31,
32]. These cell lines were generated through transient expression of Cas9 via plasmid transfection, along with introducing one or more sgRNAs targeting the gene of interest, similar to the plasmid we developed for use in
Cx. quinquefasciatus cells [
27]. In our literature review, we found only one mosquito cell line that stably expresses Cas9, the
Anopheles coluzzi-derived cell line Sua-5B-IE8-Act::Cas9-2A-Neo, made through recombination-mediated cassette exchange [
28]. The stable expression of Cas9 in mosquito cell lines can allow for large-scale genetic screening [
28]. Additionally, a stable Cas9-expressing cell line can save on reagents, as only an sgRNA would need to be introduced for genome editing.
In the present study, we developed a
Cx. quinquefasciatus-derived cell line constitutively expressing Cas9 through the introduction of a single plasmid that contained Cas9 flanked by homology arms to the
cardinal gene, based on work performed by Feng et al., 2021 [
26]. We used a combination of cell sorting and dilution into semi-solid media to generate a monoclonal line, which showed stable expression of Cas9 and successful gene editing of seven genes when single sgRNAs were introduced. We also performed a ‘mini-screen’ using sgRNAs for seven mosquito genes that we hypothesize to be either antiviral or proviral and infection with either the orthobunyavirus La Crosse virus (LACV) or the flavivirus Usutu virus (USUV). We were able to show that six of the chosen genes were antiviral in a virus-dependent manner. This new cell line will be beneficial for studying antiviral mechanisms in
Cx. quinquefasciatus.
2. Materials and Methods
2.1. Cell Culture Maintenance
The
Cx. quinquefasciatus-derived ovarian cell line (Hsu) [
33] was kindly provided by Dr. Aaron Brault (CDC for Vector-borne Diseases, Fort Collins). An Hsu-derived monoclonal cell line, named Hsu-Clone21, was generated using flow cytometry as previously described for Aag2 cells [
34]. This monoclonal cell line was used to generate the Cas9 knock-in cells. Hsu cells and any Hsu-derived cells were grown at 27 °C and 5% CO
2 in Dulbecco’s modified Eagle medium (DMEM; Corning #10-013-CV; Corning, NY, USA) supplemented with 10% FBS and 100 units/mL penicillin, 100 μg/mL streptomycin, and 5 μg/mL gentamicin.
2.2. Plasmid Transfection
To prepare cells for sorting, Hsu-Clone21 cells were seeded in 6-well plates. Hsu-Clone21 cells at 50–60% confluency were transfected with the plasmid containing vasa-Cas9 flanked by
cardinal homology regions [
26] kindly provided by Valentino Gantz and Víctor López Del Amo. GFP expression under the control of the
vasa promoter was validated in Hsu cells before the introduction of the Cas9-containing plasmid. Hsu-Clone21 cells were transfected using X-tremeGENE™ HP DNA transfection reagent (Roche, #6366236001; Pleasanton, CA, USA) as per the manufacturer’s protocol for a 6-well plate. A 2:1 DNA/transfection reagent ratio was used, and each well received 2 μg of the plasmid.
2.3. Keyence Imaging
To validate the successful transfection of the plasmid into Hsu-Clone21 cells, cells were imaged 48 h post-transfection. Aside from vasa-Cas9, the plasmid also contains dsRed under the Opie2 promoter. Prior to imaging, fresh medium was added to the cells, and images of live cells were visualized using a Keyence BZ-X710. Prior to cell sorting, Hsu-Clone21 cells transfected with the Cas9-containing plasmid were moved to 75 cm cell culture flasks (Corning, #734-0965; Corning, NY, USA). Cells were passaged for 5 weeks and imaged weekly to allow for vasa-Cas9 integration into the genome (as opposed to transient expression) prior to cell sorting.
2.4. Cell Sorting
Five weeks post-transfection, Hsu-Clone21 cells were seeded into 6-well plates. The medium was removed from Hsu cells at 70–80% confluency, and cells were washed with 2 mL of PBS. After PBS was discarded, 0.5 mL of Accumax (Thermo Fisher, #00-4666-56; Waltham, MA, USA) was added to Hsu cells, and cells were placed in the incubator for 1 min to detach cells from the plate. Cells were centrifuged in a 50 mL conical tube at 500× g for 5 min, the medium was removed, and cells were pelleted with 1 mL of sterile sorting buffer (PBS, 25 mM HEPES pH 7.0, 1% FBS heat-inactivated FBS Premium, 2 mM EDTA, 1% Pen-Strep antibiotic), with 1:10,000 DAPI to gate for living cells. Then, 1 mL of cells were filtered into 5 mL tubes through a 25 μm Strainer Cap (Olympics, #28-154; Genessee Scientific, Morrisville, NC, USA). Cells were then sorted for DAPI and dsRed expression using a BD FACSAria II Cell Sorter (Nevada Cytometry Center, Reno, NV, USA). Cells were collected in a 5 mL tube containing 1 mL of DMEM + 20% FBS and kept on ice until approximately 200,000 cells were added to each of 4 wells in a 6-well plate containing 2 mL of fresh pre-warmed DMEM, +20% FBS, and antibiotics. The medium was replaced with fresh medium 24 h post sorting and replaced daily afterward. Once cells were confluent, they were subsequently moved to a 25 cm and 75 cm flask.
2.5. Generation of Monoclonal Line Using a Semi-Solid Overlay
Semi-solid sorting was used to develop monoclonal Hsu cells stably expressing Cas9. We have observed better survival using this technique versus single-cell sorting using flow cytometry. Briefly, 45 mL of STEMCELL Technologies ClonaCell™ FLEX was added to 40 mL of 2X DMEM, 10 mL of FBS Premium Plus, and 4.44 mL of 7.5% NaHCO3. Cells previously sorted via flow cytometry (i.e., all expressing dsRed) were detached using ACCUMAX (Thermo Fisher, #00-4666-56; Waltham, MA, USA). Cells were then filtered into 5 mL tubes through a 25 μm Strainer Cap (Olympics, #28-154; Genessee Scientific, Morrisville, NC, USA), and cells were counted using a hemocytometer. The cell suspension was diluted and added to 6-well plates containing 2.5 mL of STEMCELL Technologies ClonaCell™ FLEX mixture at a concentration of 25 cells/well. Hsu cells were incubated at 27 °C in a humidified, 5% CO2 incubator. Once distinct colonies expressing dsRed formed, they were removed using a P-1000 pipette tip and transferred to a 24-well plate containing fresh DMEM, +20% FBS, and antibiotics. Cells were expanded and aliquots of cells were frozen using liquid nitrogen for storage.
2.6. PCR
To verify the integration of Cas9 into the
Cx. quinquefasciatus genome, PCR primers were designed to amplify from 252 bp outside of the right flanking homology arm within the
cardinal gene to inside the Cas9 endonuclease gene (
Table 1). Briefly, genomic DNA was extracted from our monoclonal dsRed-expressing cells as well as mock-transfected Hsu cells (negative control) using the Zymo Quick-DNA ™ Miniprep kit (Zymo Research, # D3024; Irvine, CA, USA) as per the manufacturer’s protocol. DNA was quantified using a Qubit Flex Fluorometer (Thermo Fisher, Waltham, MA, USA), normalized to 10 ng/μL. Target regions were amplified by PCR using Platinum™ SuperFi II PCR Master Mix (Thermo Fisher, #12368010 Waltham, MA, USA). DNA was quantified using a Qubit Flex Fluorometer (Thermo Fisher, Waltham, MA, USA), normalized to 10 ng/μL, and verified with gel electrophoresis.
Further, to validate Cas9 expression, RNA was extracted from our Cas9 knock-in Hsu cells five weeks after single-cell generation, as well as mock-transfected Hsu cells using the Directzol RNA miniprep kit (Zymo Research, #D4033; Irvine, CA, USA), as per the manufacturer’s protocol. RNA was quantified using a Qubit Flex Fluorometer (Thermo Fisher, Waltham, MA, USA). Following RNA extraction, cDNA was made using the High-Capacity cDNA reverse transcription kit (Thermo Fisher, #4368814, Waltham, MA, USA). PCR primers were designed to amplify a short region within Cas9 (
Table 1) to validate mRNA expression. The target region was amplified by PCR using Platinum™ SuperFi II PCR Master Mix (Thermo Fisher, #12368010 Waltham, MA, USA), and DNA was normalized to 10 ng/μL using a Qubit Flex Fluorometer (Thermo Fisher, Waltham, MA, USA), and verified through gel electrophoresis.
2.7. sgRNA Design and gRNA Transfection
CRISPR GuideXpress (
https://www.flyrnai.org/tools/fly2mosquito/web, accessed on 1 March 2021) was used to design the single guide RNAs (sgRNAs) targeting the
Cx. quinquefasciatus genomic loci of
dcr-2,
ago2b,
vago,
piwi5,
piwi6a,
spcs1, and
cullin4a (
Table 2). The sgRNAs with the highest Housden efficiency score, an off-target score of 0, and coverage of <60% were chosen and obtained by custom order from Synthego (Redwood City, CA, USA). The efficiency of these gRNAs was validated in previous experiments using transient Cas9 transfection. Monoclonal Cas9 knock-in Hsu cells were seeded into 24-well plates prior to transfection. Single gRNAs were transfected into Cas9 knock-in Hsu cells using Lipofectamine™ RNAiMAX (Thermo Fisher Scientific, #13778150; Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, 21.5 µL of Opti-MEM with 2 µL of Lipofectamine™ RNAiMAX was combined with 21.5 µL of Opti-MEM with 17.5 µM sgRNA. The lipoplex was incubated for 10 min at room temperature before addition into wells containing 0.5 mL fresh DMEM, +10% FBS, and antibiotics. Transfected cells were incubated at 27 °C and 5% CO
2.
2.8. T7 Assay
A T7 endonuclease assay was used to assess gene editing efficiency as previously described [
27,
35]. The extent of cleavage by T7 endonuclease corresponds to the degree of gene editing in the cell population. Genomic DNA (gDNA) was extracted three days post sgRNA transfection using the Quick-DNA Miniprep kit (Zymo Research). The target loci were PCR-amplified with primers flanking the edited region (
Table 3), using Q5
® Hot Start High-Fidelity Master Mix (New England Biolabs, #E2621; Ipswich, MA, USA) and 100 ng of gDNA in a 50 μL reaction. PCR products were visualized on agarose gel electrophoresis, and the bands (900–1000 bps) representing the sgRNA-target regions were isolated using a Zymoclean Gel DNA Recovery kit (Zymo Research, #D4002; Irvine, CA, USA). For the T7 endonuclease I assay, 200 ng of gel-purified PCR amplicons were mixed with 10× NEBuffer 2 (New England Biolabs, #B7002; Ipswich, MA, USA) and denatured at 95 °C for 5 min, followed by gradual cooling to form potential heteroduplexes. Then 1 μL of T7 Endonuclease I enzyme (New England Biolabs, #M030; Ipswich, MA, USA) was added, and the mixture was incubated at 37 °C for 15 min. The reaction was stopped with 1.5 μL EDTA, and the resulting products were separated by gel electrophoresis on a 1% agarose gel in parallel with the 1 kb Plus DNA ladder (New England Biolabs, #N3200; Ipswich, MA, USA).
2.9. Gene-Specific dsRNA Synthesis
Gene-specific dsRNA was synthesized using the MEGAScript™ RNAi Kit (Thermo Fisher, #AM1626, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, PCR primers containing T7 promoter sequences (
Table 4) were designed to amplify the
ago2b gene from Hsu cDNA, which was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher, #4368814, Waltham, MA, USA). For the non-specific control, GFP dsRNA was synthesized using a GFP-containing plasmid and T7 primers (
Table 1). PCR amplification of target regions was performed using Q5
® Hot Start High-Fidelity Master Mix (New England Biolabs, #M0494) and verified by gel electrophoresis. PCR products were purified and concentrated using the Zymo DNA Clean & Concentrator™-25 Kit (Zymo Research, #D4033, Irvine, CA, USA). The purified DNA was transcribed into dsRNA in vitro using the MEGAScript™ RNAi Kit (Thermo Fisher, #AM1626, Waltham, MA, USA).
2.10. Transfection of dsRNA and siRNA
Hsu cells were seeded into 24-well plates with 500 µL of culture medium per well. After overnight incubation at 27 °C to facilitate cell adhesion, transfections were performed using gene-specific dsRNA and a non-specific dsRNA control (GFP). For dcr-2 silencing we used a gene-specific siRNA (GAAGAAGUACCUUCUCUACAAGGAA) compared to a Renilla luciferase siRNA control (AGAAGUUCCCUAACACCGAGUUCGU), which were both purchased from Integrated DNA Technologies (Coralville, IA, USA) as dsiRNAs. Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher, #13778075, Waltham, MA, USA) was used as per the manufacturer’s protocol. Specifically, 1.5 µL of Lipofectamine™ RNAiMAX reagent (Thermo Fisher, #13778100, Waltham, MA, USA) was diluted in 25 µL of Opti-MEM™ Reduced-Serum Medium (Thermo Fisher, #31985062, Waltham, MA, USA) per well. Separately, 500 ng of dsRNA or 5 pmol siRNA was diluted in 25 µL of Opti-MEM™ per well. The diluted dsRNA or siRNA was then mixed with the diluted Lipofectamine reagent and incubated at room temperature for 15 min. Then, 50 µL of the resulting dsRNA-Lipofectamine complex was added to each well, followed by 450 µL of fresh complete culture medium. The cells were incubated for 48 h and then infected with either LACV or USUV.
2.11. Virus Infection
Hsu cells were seeded in 24-well plates at a density of 2 × 105 cells per well and infected with either LACV at a multiplicity of infection (MOI) of 10 or USUV at an MOI of 50. The virus was diluted in 250 µL of culture medium (DMEM or Schneider’s medium without additives) per well and added to cells after the existing complete medium was removed. Following a 2 h incubation at 27 °C, the virus-containing medium was aspirated, and 500 µL of complete culture medium was added to each well. Plates were incubated at 27 °C for 48 h, and cells were lysed as per the manufacturer’s instructions; RNA extraction was performed using the Directzol RNA miniprep kit (Zymo Research, #D4033; Irvine, CA, USA).
2.12. Quantitative Reverse-Transcriptase qRT-PCR
qRT-PCR was conducted using the primers in
Table 4 and the iTaq™ Universal SYBR
® Green One-Step Kit (Bio-Rad, #1725150, Hercules, CA, USA) or the iTaq™ Universal Probes One-Step Kit (Bio-Rad, #1725141, Hercules, CA, USA) on a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Gene or viral RNA levels were normalized to the previously validated housekeeping gene actin-5c, or virus standards were used to determine virus levels.
4. Discussion
In this study, we successfully developed a stable Cas9-expressing cell line derived from a monoclonal
Cx. quinquefasciatus Hsu cell line, providing a valuable tool to study gene function in this understudied mosquito species. Our results demonstrated Cas9 activity and gene-editing of seven genes that are of interest as candidate antiviral or proviral genes. Using these Cas9 knock-in cells, editing of the
dcr-2 locus, specifically, was more apparent compared to our previous plasmid-based transient expression of Cas9 and sgRNA [
27]. This gene editing following a simple transfection of sgRNAs into our cell line can facilitate functional studies of mosquito immune genes to understand vector competence and viral interactions. We performed such an experiment and were able to show antiviral activity for six
Cx. quinquefasciatus genes. Specifically, we showed antiviral activity for
dcr-2,
ago2b, and
vago during LACV infection and
piwi5,
piwi6a, and
cullin4a during USUV infection. To the best of our knowledge, this study is also the first gene knock-in in Hsu cells and acts as a proof-of-principle for future knock-ins of exogenous genes in this cell line, such as dCas9 [
38], dCas9-VPR [
39], and Cas13 [
40], to increase our molecular toolkit or other genes to further study antiviral responses in vitro.
Viswanatha et al., 2021, generated an
Anopheles coluzzii cell line with stable Cas9 expression using recombination-mediated cassette exchange. This cell line was used to perform large-scale CRISPR screens to study gene function [
28]. By integrating Cas9 into the genome, the authors eliminated the need for repeated transfections, which is often a limitation in transient expression systems. A similar approach could be used for the
Cx. quinquefasciatus Cas9 knock-in Hsu cell line developed in our study to understand genes related to immunity and vector competence before performing in vivo experiments in
Cx. quinquefasciatus mosquitoes.
Using seven select sgRNAs, we performed a ‘mini-screen’ and made a few interesting discoveries. We found that the key components of the RNAi machinery Dcr-2 and Ago-2 are antiviral against LACV but not USUV in Hsu cells. We observed the same result with RNAi-mediated gene silencing, but the impacts on LACV replication were more pronounced in our Cas9-mediated screen compared to RNAi, suggesting a more robust tool to identify antiviral genes. We also observed that Vago was antiviral against LACV, but not USUV. This was interesting for us to see, because Vago has previously been implicated in antiviral responses to WNV, a virus closely related to USUV. In contrast, the editing of two
piwi genes,
piwi5 and
piwi6a, had no effect on LACV replication, but significantly increased USUV replication. We previously observed a trend for antiviral activity of
piwi5 against USUV using RNAi-mediated gene silencing, but only observed a very modest increase and no statistical significance [
22]. This highlights again the increased sensitivity of this type of screen compared to RNAi in Hsu cells.
Finally, we included two genes that we thought may have proviral activity,
spcs1 and
cullin4a, based on previous studies showing that the SPCS1 protein is required for flavivirus assembly in mammalian cells [
37] and that the ubiquitin ligase Cullin4 is a proviral factor in
Cx. quinquefasciatus mosquitoes and cells during WNV infection [
36]. We observed a non-significant trend for proviral activity of SPCS1 during USUV infection, but no effect on LACV replication. Since we only measured RNA replication here and did not perform infectious virus titrations, it is possible that any proviral effect of SPCS1 would be more pronounced and statistically significant on virus titers due to its role in virus assembly [
37]. SPCS1 in mammalian cells has no antiviral or proviral effects on bunyaviruses, so it is unsurprising that LACV was not affected by
spcs1 editing. The other hypothesized proviral protein, Cullin4, is a cullin RING ubiquitin ligase that degrades the signal transducer and activator of transcription (STAT) and thus blocks immune responses associated with JAK/STAT signaling [
36]. It was shown that the silencing of
cullin4a gene expression reduced WNV replication and overexpression increased WNV replication [
36]. However, we observed an increase in USUV replication following transfection of a
cullin4a-targeting sgRNA. While unexpected, we also showed that USUV is not affected by the secreted cytokine
vago (unlike WNV), which activates JAK/STAT signaling in
Cx. quinquefasciatus mosquitoes [
21], suggesting that JAK/STAT signaling has no effect on USUV. How
Cx. quinquefasciatus Cullin4 mediates antiviral activity against USV remains unknown. Follow-up in vivo studies would be required to shed further light on the impacts of Cullin4 on USUV in vivo. Overall, our results demonstrate the use of this Cas9 knock-in cell line and how antiviral immune responses may have unique effects on different virus–vector combinations.
Using a similar workflow to the present study, future work will focus on generating a non-cleaving Cas9 (dCas9) and dCas9-VPR-expressing Hsu cell line, allowing transcriptional control of endogenous genes. Similar dCas9 systems have been utilized in
Aedes albopictus C6/36 cells and
Aedes aegypti mosquitoes as a CRISPR activation system to upregulate genes of interest [
41,
42]. In
Drosophila, dCas9 has been used for CRISPR interference, for example, to reduce the transcription of long noncoding RNAs [
43]. We have shown that the
cardinal gene serves as a site for integration and successful gene expression in Hsu cells and could be used for the knock-in of a variety of genes, including dCas9 and dCas9-VPR. Overall, developing a dCas9-based system in
Cx. quinquefasciatus-derived cells will provide a versatile approach to studying gene function and regulation.
The limitations of any gene editing or gene silencing approach include poor annotations of the
Cx. quinquefasciatus genome, genetic variation between mosquito colonies, and the resulting need to sequence genome loci of interest to ensure that there are no mismatches in sgRNA target sites. This is not unique to sgRNAs, but we have come across sequence variations (SNPs, but also indels) in our cell lines that do not match the NCBI/VectorBase-annotated sequence, resulting in sgRNA designs that cannot cleave as anticipated. However, this is also a concern when using siRNAs, which may not allow multiple mismatches to mediate gene silencing. A recent assembly of the
Culex quinquefasciatus genome may help the development of robust sgRNAs, but also highlights high variation between
Cx. quinquefasciatus genomes [
44]. Poor genome annotation may be most challenging for the future design of CRISPRa technology due to the need for sgRNAs that bind in noncoding regions (promoter regions), which may be annotated even less reliably than coding sequences. Hopefully, as molecular methodologies and research develop further, genome annotations will improve as well, but natural variation between mosquito colonies will always have to be taken into consideration.
In summary, we developed a Cas9 knock-in Hsu cell line that provides an efficient platform for genome editing. Our cell line expands the current toolkit and will facilitate the future study of Cx. quinquefasciatus immune genes and genes related to vector competence. The present study provides a new tool to facilitate the development of genetic strategies to combat mosquito-borne diseases.