1. Introduction
Coronary artery disease (CAD) is a form of cardiovascular disease caused by plaque accumulation on the walls of coronary arteries that supply blood to the heart. In atherosclerotic disease, these plaques eventually block blood flow to the heart. Because of this, CAD can lead to heart failure, a condition in which the heart’s ability to pump blood is reduced [
1]. Risk factors related to CAD include hypertension, smoking, hyperlipidemia, and diabetes [
2,
3,
4,
5]. Several studies have reported that polymorphisms in genes related to fibrin degradation and fibrin coagulation are also associated with CAD [
6,
7]. In particular, some variants in the promoter region of plasminogen activator inhibitor-1 (also known as SERPINE1 or
PAI-1), which regulates fibrinolysis, are thought to be associated with CAD sensitivity [
8,
9].
Micro (mi)RNAs are a class of small (18–22 nucleotides) non-coding RNAs that play important regulatory roles in gene expression. By binding to 3′-untranslated regions (UTRs) in target mRNAs, miRNAs regulate post-transcriptional gene expression by causing translational repression or mRNA degradation [
10,
11]. Through these mechanisms, miRNAs are involved in the regulation of cell growth, cell proliferation, apoptosis, and cell differentiation [
12]. In addition, miRNAs play essential roles in the cardiovascular system by regulating heart and blood vessel development as well as cardiovascular diseases [
13]. Recently, several miRNAs were found to regulate CAD development [
14,
15].
The
PAI-1 gene, which encodes a member of the serine protease inhibitor superfamily, is located on chromosome 7.
PAI-1 is produced by endothelial cells, platelets, and other cell types and is associated with several disease conditions including diabetes, hypertension, metabolic syndrome (MetS), and obesity. When plasminogen is converted to plasmin, the fibrinolytic system is initiated [
16]. When plasmin is activated by tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA), it causes fibrinolysis, matrix metalloprotease activation, and extracellular matrix degradation [
17]. This fibrinolytic system is regulated by
PAI-1. Studies have shown that polymorphisms in
PAI-1 are associated with CAD [
18].
The above data suggests a role for both PAI-1 and miRNAs in CAD. Based on these associations, we wondered how expression-altering miRNAs that bind to the 3′-UTR of PAI-1 impact the risk for CAD and if there are any associations between polymorphisms in these miRNA genes and other CAD risk factors. To answer these questions, we designed a genetic epidemiological study to test for associations between CAD risk in the Korean population and five polymorphisms of miRNAs binding to the 3′-UTR of the PAI-1 gene: miR-30c rs928508 A > G, miR-143 rs41291957 G > A, miR-143 rs4705342 T > C, miR-145 rs353291 T > C, and miR-181a2 rs10760371 T > G.
3. Discussion
We evaluated associations between CAD risk and five polymorphisms in miRNAs targeting the 3′-UTR of
PAI-1. Associations have been reported between these five polymorphisms and other diseases, including non-small-cell lung cancer (
miR-30c rs928508) [
19], colorectal cancer (
miR-143 rs41291957) [
20], ischemic stroke (
miR-143 rs4705342) [
21], atherosclerosis (
miR-145 rs353291) [
22], and methamphetamine addiction (
miR-181a2 rs10760371) [
23]. However, only
miR-143 rs41291957 was previously investigated in relation to CAD [
24]. Furthermore, although these SNPs have been studied in many countries, they have not been analyzed in Korea. Thus, to our knowledge, our study represents the first attempt to elucidate the effect of these five miRNA SNPs on the prevalence of CAD in Koreans.
In a prior study,
miR-34a was shown to bind to the 3′UTR of
PAI-1 and regulate
PAI-1 expression, with
miR-34a overexpression leading to reduced expression of
PAI-1 [
25]. Similarly,
miR-143, another miRNA investigated in this study, was reported to bind to the 3′UTR of
PAI-1 and inhibit its expression [
26]. Thus, published findings indicate that miRNA binding to
PAI-1 lowers the expression level of this gene.
PAI-1 encodes a protein that inhibits tPA and uPA, disrupts the fibrinolytic system, and contributes to the pathogenesis of cardiovascular disease. Accordingly, elevated expression of
PAI-1 leads to an increased risk for CAD [
27]. Based on the above, we predict that binding of miRNA to the
PAI-1 3′UTR reduces
PAI-1 expression and lowers the risk for CAD. In our study, CAD risk was significantly increased for those with the GG genotype of
miR-30c rs928508 (
Table 2). This observation suggests that individuals with GG genotype have an elevated incidence of CAD, resulting from reduced expression of
miR-30c and increased expression of
PAI-1 relative to those with the AA genotype.
CAD is a complex condition influenced by a variety of environmental and genetic risk factors, including
PAI-1 and its associated miRNAs [
28,
29,
30,
31]. Several studies have indicated a strong association between components of MetS and atherosclerotic diseases, including CAD [
32,
33]. Based on the ATP III criteria, a diagnosis of MetS is established when an individual exhibits three or more of the following five criteria: blood pressure ≥ 130/85 mmHg, waist circumference > 102 cm in males or >88 cm in females, fasting blood sugar ≥ 110 mg/dL, plasma triglyceride concentration ≥ 150 mg/dL, and plasma HDL-cholesterol concentration < 40 mg/dL in males or <50 mg/dL in females [
34]. Diabetes mellitus, another clinical characteristic, is also associated with CAD [
35,
36]. On the other hand, genetic predisposition to elevated folate levels was linked to a reduced likelihood of developing CAD [
37]. Combinations of
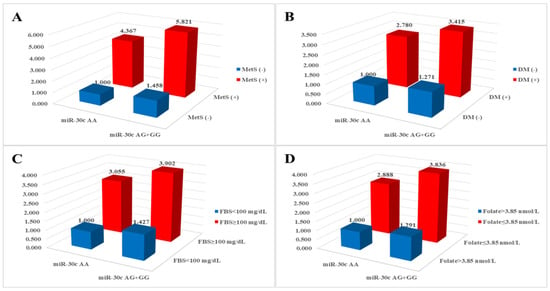
miR-30c rs928508 genotypes and risk factors associated with MetS demonstrated synergistic elevation of risk for CAD, as illustrated in
Figure 1. Subgroups characterized by the
miR-30c AG + GG genotypes and the presence of hypertension, diabetes mellitus, hyperlipidemia, BMI ≥ 25 kg/m
2, and HDL < 40 mg/dL (males) or <50 mg/dL (females) displayed significantly increased AORs (2.246, 3.415, 1.675, 1.887, and 2.008, respectively) compared with subgroups with the
miR-30c AA genotype without MetS-related conditions (
Table S2). Consistent with our findings, MetS was also shown to be an important risk factor for CAD in patients from other ethnic groups [
38,
39].
Our study has several limitations. First, although the regulatory effect of miRNA on PAI-1 is known [
25,
26], the specific mechanisms through which the five SNPs impact the development of CAD are not fully understood. Therefore, further in vitro and in vivo studies are needed to validate the effects of the SNPs on CAD risk and enhance our understanding for future applications. Second, our study sample was limited to the Korean population, which restricts the generalizability of our findings. We further note that the frequencies of these polymorphisms in the Korean population differ slightly from those in other populations, making it difficult to extend our conclusions to a global context. Consequently, it is crucial to expand our research to encompass a more diverse range of patient samples. Third, there is a dearth of information regarding other environmental risk factors for CAD.
In summary, we evaluated whether five polymorphisms of PAI-1-related miRNAs affect susceptibility to CAD in the Korean population. We found that miR-30c rs928508 was associated with CAD risk. In combination analyses, some combinations of miR-30c rs928508, miR-143 rs41291957, and miR-143 rs4705342 alleles and genotypes were also associated with increased or decreased CAD risk. Moreover, these three polymorphisms displayed synergistic effects on CAD risk when combined with clinical conditions that independently increase the risk of CAD. These findings can be used to identify new CAD prognostic biomarkers using miR-30c rs928508, miR-143 rs41291957, and miR-143 rs4705342 combined with other miRNA polymorphisms and various clinical factors.
4. Materials and Methods
4.1. Study Participants
Blood samples were obtained from 483 patients with CAD [age, mean ± standard deviation (SD) = 61.07 ± 11.46 years] and 400 age- and sex-matched healthy control participants (age, mean ± SD = 60.56 ± 11.70 years). All participants were recruited from the Department of Cardiology of CHA Bundang Medical Center, CHA University, in Seongnam, South Korea, between 2014 and 2016. All participants provided written informed consent for the study, which was approved by the Institutional Review Board of CHA Bundang Medical Center (IRB number: 2013-10-114). All study procedures adhered to the principles outlined in the Declaration of Helsinki.
The patients included in this study exhibited coronary artery stenosis of over 50% in at least one of the primary coronary arteries or their significant branches, as verified by coronary angiography. Patients with a history of cardiac arrest and a life expectancy of less than one year were excluded to mitigate the confounding effects of diverse medical interventions on blood testing. Diagnoses were established via coronary angiography and were corroborated by at least one proficient cardiologist. The 400 control participants were seen at the Department of Cardiology at the CHA Bundang Medical Center for a thorough health assessment, which included biochemical testing and cardiological examination. Individuals with a history of angina or myocardial infarction, as well as those exhibiting T wave inversion on electrocardiography, were excluded from the control cohort.
Hypertension was characterized by systolic pressure ≥ 130 mmHg and diastolic pressure ≥ 80 mmHg and was considered present in individuals using anti-hypertensive medications [
40]. Diabetes mellitus was defined as a fasting plasma glucose level ≥ 110 mg/dL and was considered present in individuals taking medications for diabetes. Hyperlipidemia was defined as fasting serum total cholesterol (TC) ≥ 150 mg/dL or a history of treatment with anti-hyperlipidemic agents. Smoking status referred to individuals currently engaged in smoking [
18].
4.2. Blood Biochemical Analyses
Blood (2 mL) was obtained in tubes containing anticoagulant after a 12 h fasting period. To isolate plasma from whole blood, the samples were centrifuged at 1000× g for 15 min. Plasma concentrations of homocysteine and folate were measured using the IMx fluorescence polarizing immunoassay (Abbott Laboratories, Abbott Park, IL, USA) and a radioimmunoassay kit (ACS:180; Bayer, Tarrytown, NY, USA), respectively. The levels of total cholesterol, triglycerides, HDL-cholesterol, and LDL-cholesterol were determined by colorimetric enzymatic methods using commercial reagent sets (TBA 200FR NEO, Toshiba Medical Systems, Otawara, Japan).
4.3. SNP Selection
To identify miRNAs that bind to
PAI-1 mRNA, an online search was performed using TargetScan (
http://www.targetscan.org, accessed on 5 September 2023) (
Figure S1) and TarBase v8 databases (DIANA tools—TarBase v8 (uth.gr), accessed on 5 September 2023) (
Figure S2). The targeted mRNA sequences, primarily located in the 3′-UTR, were acquired from the National Center for Biotechnology Information (NCBI;
www.ncbi.nlm.nih.gov/, accessed on 5 September 2023). We first selected overlapping SNPs from the results of an online search. We then searched the SNPs in NCBI and selected only those found in East Asian populations and with an alternative allele frequency of ≥0.05.
4.4. Genetic Analyses
DNA was isolated from white blood cells in peripheral blood using the G-dex II Genomic DNA Extraction kit (iNtRON Biotechnology, Inc., Seongnam, Republic of Korea), following the guidelines provided by the manufacturer. After extraction, the quantity (A260) and quality (A260/A280 ratio) of genomic DNA were promptly evaluated with a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Genomic DNA purity was further confirmed by analyzing the patterns of DNA fragments obtained from agarose gel electrophoresis.
miR-30c rs928508 A > G,
miR-143 rs41291957 G > A,
miR-143 rs4705342 T > C,
miR-145 rs353291 T > C, and
miR-181a2 rs10760371 T > G were genotyped using a TaqMan™ SNP Genotyping Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with a Rotor-Gene 6000 Real-Time PCR System (QIAGEN, Hilden, Germany). A solution was prepared for real-time PCR by combining 1 µL genomic DNA (100 ng/µL), 7.5 µL TaqMan Genotyping Master Mix (Applied Biosystems, Waltham, MA, USA), 0.75 µL TaqMan SNP Genotyping Assay (Applied Biosystems), and distilled water to achieve a final volume of 15 µL. The experimental procedure included appropriate negative controls. The thermal cycling conditions for this experiment were as follows: initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 1 min [
41]. To confirm the genotypes, 30% of the samples for each polymorphism were randomly selected and subjected to base sequence analysis using an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The sequencing results were 100% consistent with the genotyping results. Primer sequences for the five SNPs are listed in
Table S3.
4.5. Statistical Analysis
To compare clinical characteristics between the controls and the patients with CAD, Chi-square tests and Student’s
t-tests were used for categorical data and continuous data, respectively. Logistic regression was used to estimate associations between the polymorphisms and CAD risk. The AORs were adjusted by age, sex, hypertension, diabetes mellitus, hyperlipidemia, and smoking status. Genotyping was performed by treating the most common homozygous genotype as the dominant model and the less common homozygous genotype as the recessive model. To minimize false positives in the results, a false discovery rate (FDR) correction was applied to the
p-values using the formula q =
p * (n/k), where n represents the total number of
p-values and k indicates the rank of the
p-values when arranged from smallest to largest [
42]. One-way analysis of variance was performed to investigate the differences in various clinical factors depending on the genotypes of the polymorphisms. Statistical significance was accepted at the
p < 0.05 level. The allele frequencies obtained from each cohort were calculated to assess the adherence or departure from HWE. Analyses were performed using GraphPad Prism 4.0 (GraphPad Software Inc., San Diego, CA, USA) and Medcalc version 12.7.1.0 (Medcalc Software, Mariakerke, Belgium). Haplotypes for multiple loci were estimated using the expectation–maximization algorithm with SNPAlyze (version 5.1; DYNACOM Co, Ltd., Yokohama, Japan).