1. Introduction
Peas (
Pisum sativum L.) are a globally significant legume that play a vital role in human nutrition and serve as a protein-rich feed for livestock. Despite their importance, genomic research focusing on peas remains limited due to the complexity of their genetic makeup. However, recent breakthroughs in genomic technologies, such as the creation of genetic resource databases, the advent of high-throughput genotyping techniques, and the assembly of reference genomes, have opened new avenues for pea genomics [
1]. These advances have not only shed light on the domestication and breeding history of peas but have also assisted in the discovery of genes linked to key traits [
2]. For instance, the sequencing of the pea genome has marked a significant leap forward in our theoretical understanding with practical applications in plant breeding [
3]. Historically, the European Union has been a major producer of peas, accounting for a substantial portion of the global production. With the anticipated rise in demand for vegetable protein by 2035, the significance of the global pea market is expected to expand further [
4]. Yet, the exploration of pea genomics and gene functionality, particularly in the context of seed nutrition and abiotic stress responses, continues to present scientific challenges.
The enzyme
trehalose-6-phosphate synthase (
TPS) plays an integral role in pea seed development [
5]. It catalyzes the synthesis of
trehalose-6-phosphate (
T6P), a critical signaling and regulatory molecule implicated in plant growth, development, and stress responses [
6]. Furthermore, it regulates sucrose levels, impacts its synthesis, and aligns the growth of sink organs with the availability of sucrose [
7]. There is a pressing need to unravel the significance of TPS in seed development in order to enhance our comprehension of peas as a crop and to develop more effective breeding and seed production strategies.
In the realm of abiotic stress responses,
TPS genes have emerged as pivotal in various plant species. For instance, in sesame, quinoa, soybean, rapeseed, and pepper,
TPS gene expression is significantly modulated under different stress conditions, such as drought, salt stress, and osmotic stress, highlighting their contribution to stress adaptations [
8,
9,
10,
11,
12]. The
SiTPS10 gene in sesame, for example, is upregulated under multiple stress conditions, suggesting its key role in stress tolerance [
8]. In rapeseed,
ArTPS genes are implicated in the response to NaCl stress, thereby influencing the metabolism of polysaccharides and glycosides and the accumulation of glycyrrhizin [
11]. Despite similar findings in other plants, the specific functions of the
TPS genes and their involvement in stress responses in peas remain elusive. Given the agronomic importance of peas and the potential of
TPS genes to enhance their resilience to environmental stressors, a detailed examination of these genes in peas is both timely and necessary.
This study leveraged cutting-edge genomic data and bioinformatic tools to identify and characterize 20 PsTPS genes in wheat. Phylogenetic analysis, gene structure examination, conserved domain analysis, and interaction network predictions were performed to explore the evolutionary and structural attributes of PsTPSs. Additionally, the expression profiles of PsTPSs were systematically investigated across 20 RNA-seq samples, with a select group of hormone-responsive candidates further validated via transcriptome sequencing analysis. This study lays down a theoretical reference for future functional studies on PsTPSs and elucidates their roles in hormone responses and plant growth regulation.
2. Materials and Methods
2.1. Identification and Sequence Analysis of TPS Genes from Pisum sativum
We procured the complete genomic sequence of the peas from the National Center for Biotechnology Information (NCBI,
https://www.ncbi.nlm.nih.gov/, (accessed on 18 February 2024)) database. Utilizing a collection of
trehalose-6-phosphate synthase (
TPS) protein sequences from diverse species, also retrieved from NCBI (GCA_024323335.1), we formulated a hidden Markov model (HMM). This model served as the foundation for deploying the HMMER software suite (v3.3.2) in a search against the peas local protein database, applying a stringent E-value cutoff of 10
−5 to filter the results. The process resulted in the compilation of a preliminary roster of
TPS candidate sequences, which were subsequently curated to eliminate any duplicates. In rice,
Arabidopsis, and soybean, the HMMER model construction was also used [
13,
14].
To substantiate the authenticity of these candidate sequences, we engaged the NCBI Conserved Domain Database (CDD) and the SMART tool, both employing an E-value threshold of 10−5 and employing filters that targeted the TPS domain sequence. The TPS genes that passed this validation were designated new names which were reflective of their chromosomal coordinates within the pea genome.
For the prediction of the subcellular destinations of TPS proteins, we deployed the Cell-PLoc software (v2.0). Concurrently, the ExPASy ProtParam tool was harnessed to forecast a spectrum of physicochemical properties of the proteins, encompassing molecular weight (MW), isoelectric point (pI), instability index, and grand average of hydropathicity (GRAVY), providing a comprehensive profile of the protein characteristics of each TPS.
2.2. Phylogenetic Relationship, Gene Structure and Conserved Motifs Analysis
To elucidate the phylogenetic ties among the TPS genes of peas, a series of multi-sequence alignments were executed on the recognized TPS proteins from the peas, alongside those from Manihot esculenta, Populus trichocarpa, and Arabidopsis. The ClustalW software (Version 2.1) was the instrument of choice for these alignments. Subsequently, the sequences were subjected to IQ-TREE 2, which applied the maximum likelihood (ML) method to construct an evolutionary tree, reinforced with a bootstrap value of 1000 for statistical support.
The precise chromosomal locales and the exon–intron architectures of the
TPS genes under scrutiny were sourced from the annotated genome files, obtained from the Ensembl Plants database (
http://plants.ensembl.org/index.html, (accessed on 19 February 2024)). The physical mapping of the
TPS genes onto their respective chromosomes was accomplished through MapGene2Chromosome v2.0 (
http://mg2c.iask.in/mg2c_v2.0/, (accessed on 20 February 2024)), while the exon–intron composition was delineated using the Gene Structure Display Server (GSDS2.0) (
http://gsds.cbi.pku.edu.cn/, (accessed on 20 February 2024)), an online tool adept at such visualizations.
In addition to the structural and localization analyses, the MEME suite was engaged to forecast the conserved motifs within the sequence of each TPS protein. The search parameters were set to identify a maximum of 15 motifs, with all other settings adhering to the default configurations of the MEME tool (
http://meme-suite.org/, (accessed on 20 February 2024)).
2.3. Chromosomal Distribution, Syntenic Analysis, and Predicting the Protein–Protein Interaction Network of the PsTPSs
The downstream analysis function in MCScanX (Version 2.0) was used to calculate the Ka and Ks of the segmental and tandem duplicate gene pairs. The Ks values were used to calculate the dates of duplication events (T) according to the following equation: T = Ks/2λ, with λ = 1.5 × 10
−8 s for dicots [
15]. The Ka/Ks value was further employed to identify the selection mode of the
PsTPSs. STRING (
https://string-db.org/, (accessed on 20 February 2024)) was used to construct the functional interaction network of the proteins.
The positional and structural details of genes mapped onto the chromosomes of pea were sourced from the GFF3 file, which was obtained from the peas genome database (
https://www.peagdb.com/download/, (accessed on 20 February 2024)). Subsequently, theMapGene2Chrom web v2 (
http://mg2c.iask.in/mg2c_v2.0/, (accessed on 20 February 2024)) was engaged to graphically situate the
PsTPS genes onto their corresponding chromosomes. An examination of the syntenic relationships among the
TPS genes in pea,
Arabidopsis thaliana, and
Glycine max was performed, with the findings visualized using the Multiple Collinearity Scan Toolkit (MCScanX) and TBtools software (
https://bio.tools/tbtools, (accessed on 20 February 2024)). The genomic datasets for
Arabidopsis thaliana and
Glycine max were extracted from the Phytozome12 database (
https://phytozome.jgi.doe.gov/pz/portal.html, (accessed on 22 February 2024)).
2.4. Material and Treatments
The cultivation of the TM-1 seeds of the ‘Zhewan No.1’ (ZW1) cotton variety commenced in a fertile matrix of soil within a controlled-environment greenhouse. The greenhouse was programmed to offer a 16 h photoperiod at a constant temperature of 27 °C, succeeded by an 8 h period of darkness at 22 °C, with relative humidity levels sustained between 60% and 80%. Once the TM-1 pea seedlings had developed to a stage marked by the emergence of three leaves, and exhibiting signs of vigorous health and consistent growth, they were subjected to experimental conditions aimed at eliciting responses to drought and saline stress.
To elicit salt stress, the root systems of the seedlings were irrigated with a solution containing 300 mM of sodium chloride (NaCl). Conversely, to simulate drought conditions, the roots were treated with a 20% solution of polyethylene glycol 6000 (PEG6000). Three-leaf-stage pea (Pisum sativum L.) seedlings were cultivated in a 70% vermiculite and 30% peat moss mixture. Group 1 received a 300 mM NaCl solution for 24 h to induce salinity stress. Group 2 was treated with a 20% PEG6000 and 300 mM NaCl solution for 24 h to simulate drought stress. A control group was watered normally. Leaf samples were collected at 0 and 24 h post treatment for further analysis. Each leaf sample was promptly submerged in liquid nitrogen for flash-freezing and then securely preserved at −80 °C, ensuring the integrity of the RNA for subsequent RNA extraction and experimental analysis.
2.5. Transcriptome Sequencing
After the treatment at both the 3 h and 24 h marks, the pea seedling leaves from both the control group without treatment and those treated with 300 mmol NaCl for the respective durations, along with leaves treated with PEG6000, were rapidly frozen in liquid nitrogen and sent to Tianjin Jizhi Gene Technology Co., Ltd. (Tianjin, China) for advanced analysis. To ensure experimental reliability, three biological replicates were performed. The total RNA from these leaf samples was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The RNA integrity was evaluated by agarose gel electrophoresis, where the total RNA exhibited clear 28S and 18S rRNA bands with the 28S band approximately twice as intense as the 18S band. Subsequently, the samples were sequenced using the Illumina Novaseq 6000 platform. Library construction was performed in accordance with the protocols provided in the Illumina TruSeq RNA Sample Prep Kit. The RNA sequencing libraries were prepared through a series of steps including total RNA extraction, mRNA enrichment and fragmentation, cDNA synthesis, double-stranded cDNA end-repair, adapter ligation, and PCR enrichment.
The raw sequencing reads were subjected to a rigorous quality control and were cleaned to ensure high quality. These refined reads were then aligned to the reference genome, accessible through the specified link, to assemble transcripts and measure gene expression levels. The quality of the RNA-seq alignments was carefully reviewed. The RNA-seq data were analyzed using Weighted Gene Co-expression Network Analysis (WGCNA) to assess and standardize the transcriptomic data quality. The subsequent transcriptome analysis was conducted on a specialized cloud-based platform developed by Tianjin Jizhi Gene Technology Co., Ltd., ensuring a thorough and detailed examination of the data.
4. Discussion
Trehalose-6-phosphate synthase (
TPS) genes are integral components of plant biochemistry, serving as critical catalysts for the synthesis of trehalose, a key component in the defense mechanisms of a plant against environmental stress [
8]. Trehalose, characterized by its non-reducing disaccharide structure, is recognized for its protective role against a variety of stressors in living organisms [
8]. In
Arabidopsis, 11
TPS genes with TPS and TPP-like domains were identified, including the active AtTPS1, revealing the role of the TPS family in plant physiology and abiotic stress responses [
16]. Thirty-one
ScTPS genes were identified in sugar cane, exhibiting differential expression in response to simulated drought, salinity, and ABA stress [
17]. References are placed in the appendix. Within the plant kingdom,
TPS genes are not only pivotal for the synthesis of trehalose but also play a responsive role under stress conditions, such as those induced by high salinity [
8]. They also hold significant implications in plant metabolic pathways, including those related to glycometabolism and the management of polysaccharides and glycosides [
8]. Of note, these genes have been discovered and described across a spectrum of plant species, spanning from sesame to rapeseed, quinoa, and soybean [
5,
9,
10], and are distributed across various chromosomes with potentially diverse cellular locations. Through phylogenetic evaluation, gene structural analysis, and the identification of conserved motifs,
TPS genes have been classified into different categories. Expression studies have unveiled patterns of tissue specificity and reactivity to stress, with certain genes, such as
SiTPS10 in sesame and
BnTPSs in rapeseed, showing potential in optimizing plant resilience against a range of abiotic stresses [
8]. In summary,
TPS is a fundamental factor in the regulation of plant growth, development, and adaptation to environmental stressors.
Our research offers an in-depth examination of the diversity in structure and function of the Trehalose-6-phosphate synthase (TPS) genes within peas. It emphasizes the preservation of characteristics within TPS subfamilies and the divergence observed across different species while also elucidating gene duplication and the broader evolutionary context. Through our investigation, 20 pea genes responsible for encoding Trehalose-6-phosphate synthase were identified and categorized into four subfamilies using phylogenetic analysis. The findings exposed both similarities and differences in TPS genes compared with other plant species, indicating a diversification in structure and function across the examined plant species.
The analysis of motifs and gene structures revealed that TPS genes with close phylogenetic relationships were more likely to have analogous motif compositions and exon–intron arrangements, a pattern also noted in species such as quinoa and Brassica napus L. A significant variation in the count of exons and introns was observed in PsTPSs, with numbers ranging from 0 to 17, echoing the variability observed in species such as Brassica rapa L. Meanwhile, protein homology modeling demonstrated that genes within the same cluster share similar secondary and three-dimensional protein structures, whereas those from different subfamilies display a range of structural profiles. This conservation and diversity within subfamilies is consistent with the outcomes of phylogenetic analysis and classification. The structural variations among PsTPSs are likely to contribute to the functional diversity within the TPS gene family.
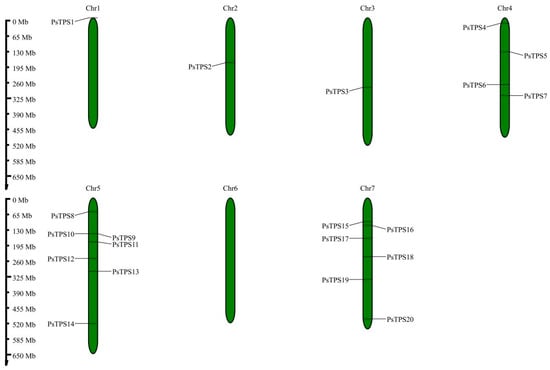
Our study also identified two gene pairs within the pea
TPS gene families, indicative of gene duplication events, with both pairs being segmental duplicates. This suggests that the
TPS gene family in peas may have expanded primarily through segmental duplication mechanisms. Homologous genes between peas and other species were localized to specific chromosomes, namely
Ps1,
Ps2,
Ps4,
Ps5, and
Ps7, which have been conserved throughout the evolutionary history of peas. These findings suggest that changes in gene structure, potentially due to the gain or loss of exons or introns, could lead to functional differences among
TPS genes [
18]. This insight into the evolutionary trajectory and structural plasticity of
TPS genes in peas contributes to our understanding of their potential roles in adaptation and diversification.
The regulatory roles of the cis-elements of the
PsTPS gene family are central to the multifaceted processes of plant growth, development, and adaptation to environmental conditions. Examination of the promoter regions of the
PsTPS genes predicted the presence of various cis-acting elements, which can be broadly divided into three categories: developmental response elements, hormone response elements, and stress response elements. Among them, elements that respond to light were the most abundant, highlighting the critical regulatory influence of light on gene expression. The characteristics of light, such as quality, intensity, and duration, exert a substantial impact on plant physiological processes such as photomorphogenesis, flowering, fruit pigmentation, and secondary metabolism [
19,
20]. Moreover, light has been established as influencing plant responses to various biotic and abiotic stresses, including photoinhibition and the management of stress response pathways [
21]. A comprehensive understanding of the light-responsive elements and their interactions with other signaling pathways is essential for promoting plant growth, development, and stress recovery.
As is well documented, hormone response elements play a cardinal role in the hormonal signaling system of plants, located in the regulatory regions of target genes and interacting with transcription factors activated by plant hormones [
22]. These interactions modulate gene expression which is critical for the plant growth and development processes regulated by hormones. Stress response elements are implicated in the adaptive mechanisms of plants to abiotic stresses such as drought and salinity, which can markedly affect the productivity of economically valuable plants [
23]. The
PsTPS gene family includes members with cis-acting regulatory elements associated with the response of plants to abiotic stress, playing a crucial role in environmental adaptation by triggering the necessary physiological and molecular responses to mitigate stress [
24,
25]. The distribution and proportion of these cis-regulatory elements vary among the
PsTPS subfamilies, suggesting functional specialization.
Transcriptome technology was used to examine the expression profiles of PsTPS genes under various abiotic stress conditions. The analysis revealed that members of the same subfamily exhibited unique expression profiles, indicative of functional diversification. Under simulated drought conditions, significant differences in the expression levels of the 14 monitored PsTPS genes were observed at multiple time points compared to the baseline (0 h). The expression levels of the two genes also displayed significant changes at different time points following the salt stress treatment compared to the control group (0 h). These findings collectively imply that PsTPS genes are integral to the regulatory network that mediates the plant’s response to drought and salt stress.
The exploration of the PsTPS gene family yielded valuable insights into the genetic mechanisms employed by plants to adapt to stress. Leveraging this understanding is crucial for the strategic modification of these genes, with the aim of developing crop varieties that possess superior resilience to environmental stressors. Exploring the PsTPS gene family uncovered potential targets for genetic enhancement, thereby paving the way for crops that can withstand the challenges posed by adverse environmental conditions. This targeted approach to crop improvement is anticipated to significantly bolster agricultural productivity and sustainability amidst climate variability and other environmental challenges.