1. Introduction
Eels are among the most prominent species in the inland aquaculture industry of South Korea. Between 2012 and 2021, eel production increased by approximately 3.6-fold, reaching 15,764 tons in 2021, accounting for approximately 47% of the total inland aquaculture output [
1]. In South Korea, most eels are reared in recirculating aquaculture systems (RASs), where water temperatures are maintained between 27 and 32 °C throughout the year, even during winter [
2]. Although this stable environment optimizes eel growth, it poses significant risks. Specifically, in the event of a disease outbreak, the shared filtration systems in RASs can facilitate rapid disease transmission across tanks, potentially leading to mass mortality and substantial economic losses. Therefore, early detection and rapid response to disease outbreaks are critical to mitigating these risks.
Recent studies have reported mass mortality in cultured eel species in South Korea, including
Anguilla japonica,
A. bicolor, and
A. marmorata, due to infections by two DNA viruses: Anguillid herpesvirus 1 (AnHV) and Japanese eel endothelial cells-infecting virus (JEECV) [
3,
4,
5,
6]. In eel farms, disease outbreaks caused by these viruses typically occur one month after stocking, leading to approximately 50% mortality within three months. In particular, their co-infection has been reported to result in mortality rates as high as 90%, highlighting the severe economic losses experienced by eel farms [
3,
4,
5,
6]. JEECV, the causative agent of viral endothelial cell necrosis of eel (VECNE), is characterized by symptoms such as head reddening, pectoral fin necrosis, and gill congestion [
5,
7,
8]. AnHV, first isolated in
A. japonica and
A. anguilla in Japan in 1985 [
9], is associated with disease outbreaks in farmed eels in Japan, Taiwan, and South Korea, and typically causes symptoms such as snout reddening, skin hemorrhages, and necrosis of the skin and gills [
3,
4,
6,
10,
11,
12,
13]. Although they can infect eels independently, AnHV and JEECV often occur in conjunction with bacterial co-infections. They can also present as dual infections, resulting in more severe diseases and elevated mortality rates [
3]. Notably, a recent survey of eel farms detected JEECV in 49% of cases, AnHV in 27%, and co-infections in 17%, highlighting their critical prevalence in aquaculture [
3]. Until now, conventional PCR (cPCR) methods have been used to diagnose the two viruses separately [
8,
14]. However, this approach is time-consuming and thus is not practical for large-scale sample testing or urgent disease diagnosis. To address this challenge, a more rapid and high-throughput diagnostic method must be developed to improve disease management in eel farming. Moreover, developing highly sensitive diagnostic tools is crucial for enabling timely identification and response to infections.
In the present study, we aimed to establish a duplex quantitative PCR (qPCR) method utilizing hydrolysis probes for the simultaneous detection of AnHV and JEECV. We validated the efficacy of this diagnostic approach through quantitative analyses of viral presences in water and eel tissue samples from aquaculture farms. This study demonstrates the potential of this approach for early detection and improved disease control in eel farming operations.
2. Materials and Methods
2.1. Fish Sampling
Twenty-eight eels exhibiting symptoms of viral infection were collected from aquaculture farms. Viral DNA was extracted from pooled spleen and kidney samples using the DNA Isolation Kit for Cells and Tissues (Roche, Basel, Switzerland), following the manufacturer’s instructions. DNA concentration and purity were assessed with a NanoVue™ Plus Spectrophotometer (GE Healthcare, Chicago, IL, USA). The DNA was diluted to 20 ng/μL, stored at −20 °C, and used for sequencing target genes and verifying the qPCR markers developed in this study.
2.2. Sequencing of Target Genes
Analysis of the GenBank database revealed that the DNA polymerase catalytic subunit gene (
Pol) of AnHV and the polyomavirus large T-antigen-like protein gene (
Ltlg)of JEECV had the most abundant sequence information. Using these regions from AnHV (GenBank accession number: KX027736) and JEECV (NC_015123) as reference sequences, new PCR primers were designed (
Table 1). PCR amplification was performed using these primers on viral DNA from the 28 eel specimens. PCR-positive samples were sequenced, and their
Pol and
Ltlg sequences were analyzed. The newly sequenced genes were deposited in GenBank (accession numbers: PV131738–131754) and aligned with corresponding sequences registered in GenBank. Molecular phylogenetic trees were constructed using the neighbor-joining method to establish phylogenetic relationships.
2.3. Design and Verification of qPCR Markers
The DNA sequences of AnHV registered in GenBank and the newly sequenced 13 AnHV-positive samples were aligned to identify a conserved region present in both AnHV-1 and AnHV-2. This conserved sequence was used to design highly specific qPCR primers and hydrolysis probes. The AnHV probe employed Cy5 as the fluorescent reporter dye and BHQ2 as the quencher (
Table 2). The optimal annealing/extension temperature for the AnHV qPCR was determined by testing temperatures in 2 °C increments between 60 °C and 70 °C to identify the condition that yielded the highest amplification efficiency.
Similarly, the DNA sequences of JEECV in GenBank and the newly four JEECV-positive samples were aligned to identify a conserved region. Specific qPCR primers and hydrolysis probes were designed for JEECV detection, using HEX as the fluorescent reporter dye and BHQ1 as the quencher (
Table 2). The annealing/extension temperature for JEECV qPCR amplification was optimized using the same temperature range (60–70 °C) to achieve maximum amplification efficiency.
2.4. Optimization of qPCR Conditions
To optimize the duplex qPCR conditions for detecting AnHV and JEECV, primer and hydrolysis probe concentrations were tested in the range of 5–10 pmol/rxn. The annealing/extension temperatures were set at 62 and 63 °C for the final verification. Detailed information on the reaction mixture and amplification conditions is provided in
Table 3 and
Table 4.
2.5. Standard Curves
Internal standards were established for the target genes (Pol for AnHV and Ltlg for JEECV). DNA fragments containing the forward and reverse primers, as well as hydrolysis probe binding sites, were synthesized and inserted into plasmids for mass production (Bioneer, Daejeon, South Korea). The plasmid DNA was quantified, and copy numbers were standardized to 100,000. Serial 10-fold dilutions were performed to establish standard materials with concentrations ranging from 100,000 copies/rxnto 1 copy/rxn. Calibration curves were generated for each virus to enable precise quantification of the target genes.
2.6. Sensitivity Tests
To evaluate the detection sensitivity of the diagnostic method developed in this study compared to cPCR, both cPCR and qPCR analyses were performed on 28 eel tissue samples, along with two no-template controls. All DNA samples were diluted to a concentration of 20 μg/μL, and 2 μL (40 μg) of each sample was used for the analyses. For cPCR, the primers newly designed in this study for detecting AnHV or JEECV were used (
Table 1). For JEECV detection, as the virus was not identified in the first round of PCR, nested PCR was performed to enhance sensitivity.
2.7. Spike Tests Using Culture Water
To simulate virus detection in aquaculture water, JEECV-infected eel tissues (kidney and spleen pool) were diluted 10-fold in Minimum essential medium (MEM) and completely homogenized. Subsequently, 350 μL (containing 35 mg of tissue) of eel tissue homogenate was added to 20 L of groundwater used in eel farming and mixed thoroughly. The spiked groundwater was divided into portions of 1, 2, 4, and 10 L and filtered through cellulose nitrate membranes (50 mm diameter, 0.45 μm pore size; Whatman®, Maidstone, UK). Viral DNA was then extracted using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany), following the complete destruction of the membranes. This destruction was achieved by adding three stainless steel beads (2.4 mm diameter; OMNI International, Kennesaw, GA, USA) and processing the sample with a Bead Ruptor Elite homogenizer (OMNI International).
2.8. Validation of the qPCR Diagnostic Method for Monitoring Viral Outbreaks in Eel Farms
The qPCR diagnostic method developed in this study was validated by monitoring virus occurrences in eel tissues and culture water from two eel farms monthly. One liter of rearing water was collected from aquaculture tanks and transported to the laboratory. Suspended solids were removed, and the water was filtered through a nitrocellulose membrane filter (0.45 μm pore size). Subsequently, environmental DNA (eDNA) was extracted from the membrane filter using the DNeasy Blood & Tissue Kit (QIAGEN). To monitor virus presences in eel tissues, three eels were sampled from the same tanks where water was collected. Tissues were collected from the pectoral fins, gills, kidneys, and spleen. Genomic DNA was extracted from each tissue using the DNA Isolation Kit for Cells and Tissues (Roche) and eluted in 50 μL of TE buffer (10 mM Tris-HCl, pH 8.0; 1 mM EDTA, pH 8.0). Subsequently, DNA concentration and purity were determined using a spectrophotometer. The extracted eDNA from water samples and genomic DNA from tissue samples were used as templates for quantifying viral gene copy numbers using the qPCR method
4. Discussion
In a previous study, we investigated the infection status of AnHV and JEECV in eel farms across South Korea [
3], confirming that they are widespread in cultured eels, with frequent cases of co-infection. Such mixed infections, particularly during the juvenile stage, often led to mass mortality, resulting in significant economic losses. These findings emphasize the need for monitoring both viruses to improve disease surveillance in eel farms. However, the currently available PCR methods [
8,
14] require individual detection of each virus, making the process labor-intensive and time-consuming. To address these limitations, in the present study, we developed a highly sensitive duplex qPCR method capable of detecting both viruses simultaneously.
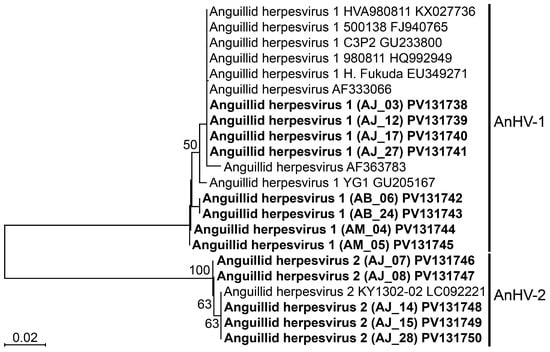
We conducted DNA sequencing of PCR-positive samples after designing primers specific genes of
Pol and
Ltlg to JEECV and AnHV, respectively. While the sequencing results revealed no genetic variation in JEECV, two types of AnHV (AnHV-1 and AnHV-2) were detected, independent of host species or geographic region. AnHV-1 has been documented in various eel species, including
A. anguilla,
A. japonica,
A. bicolor,
A. marmorata, and
A. rostrata, across Europe, East Asia, and Southeast Asia [
6,
15,
16,
17,
18,
19], with full genome sequences reported for strains from
A. anguilla and
A. rostrata [
20,
21]. AnHV-2, first isolated from
A. japonica, has been partially sequenced for
Pol (GenBank accession number: LC092221) serving as a reference. While Kim et al. have recently developed a duplex PCR method for detecting AnHV-1 [
22], in this study, we designed primers and probes targeting conserved regions of the
Pol gene shared by both AnHV-1 and AnHV-2. This approach enabled us to simultaneously detect both AnHV types. Moreover, standard curve analysis using plasmid DNA demonstrated high reliability (
r2 = 0.999) in detecting both viruses down to a single copy, confirming the exceptional sensitivity of our method for detecting trace viral loads. In addition, compared to cPCR, the duplex qPCR method developed in this study was 1.7 times more sensitive for AnHV and 2.5 times more sensitive for JEECV. The ability of our method to detect low viral loads undetectable by cPCR significantly enhances the accuracy of disease monitoring. This heightened sensitivity enables the detection of viral infections at preclinical stages, prior to the onset of symptoms, thereby facilitating early diagnosis and intervention for AnHV and JEECV in farmed eels.
In addition, using this diagnostic tool, we conducted a six-month monthly monitoring program at two eel farms and analyzed samples from rearing water and fish. We observed no cases of co-infection during the monitoring period; however, we detected AnHV and JEECV individually. At Farm A, initial investigations revealed no detectable viral DNA in either eel tissues or eDNA, suggesting either pre-infection conditions or a viral load too low to detect. In subsequent investigations, JEECV was detected in eDNA at a concentration of 1.0 × 101 copies/mL, indicating initial viral introduction. By the third investigation, JEECV was detected in all eel tissues, coinciding with an increase in eel mortality. The highest viral concentration (4.0 × 103 copies/μL) was observed in the gills, suggesting that this tissue is the primary entry point and reservoir for infection. At Farm B, AnHV was initially detected in eDNA from rearing water at concentrations below the limit of quantification (< 1 copy/mL), indicative of latent or low-level infection. In the second survey, the virus was undetectable in eDNA; however, it was present in all eel tissues, highlighting the importance of tissue sampling for identifying subclinical infections that may be missed by eDNA analysis alone. During the third survey, the virus was not detected, despite deterioration in eel health. By the fourth survey, AnHV was detected in all samples, and mortality rates had increased, marking the onset of an outbreak. These findings emphasize the necessity of continuous monitoring and early intervention to prevent mass mortality from viral infections.
The present study suggests that an integrated sampling approach, combining rearing water eDNA analysis with fish tissue examination, may provide a more accurate prediction of viral disease outbreaks than an individual approach. However, repeated sampling of live fish can impose significant stress on the stock. Therefore, continuous monitoring of rearing water eDNA using the high-sensitivity detection technology developed in this study offers an efficient and non-invasive alternative for viral disease surveillance and control. Moreover, when the timing of virus detection in stock water was compared to the onset of eel mortality, viruses were consistently detected in eDNA from three weeks to three months before mortalities were observed (
Table 7). This finding demonstrates that monitoring eDNA in rearing water alone is sufficient to predict disease outbreaks. Similarly, recent studies have highlighted the potential of eDNA methods for detecting fish viruses [
23,
24,
25,
26]. Notably, DNA viruses are more stable than RNA ones, making them particularly suitable for eDNA-based monitoring [
27]. Given that they are DNA viruses, both AnHV and JEECV serve as ideal candidates for detection using this method in aquaculture settings. Moreover, the highly sensitive eDNA detection technology developed in this study can be applied to the surveillance of other aquatic pathogens in various aquaculture systems and natural waters as well as in eel farms. This approach may be useful for the early detection and control of various diseases affecting fish, thereby contributing to improving aquatic animal health management on a broader scale.
This study has some limitations that should be considered. First, the data were collected from only two eel farms, which may not fully represent the situation across all aquaculture farms. Expanding the sample size in future research would improve the generalizability of the findings. Second, environmental factors such as water temperature, quality, and stocking density, which are known to influence viral outbreaks, were not included in the analysis. Future studies incorporating these factors could provide more comprehensive insights into disease dynamics. Third, while we used filtration methods to detect viruses in rearing water, further comparative studies exploring alternative concentration methods, such as ultracentrifugation, may enhance the detection efficiency and reliability of viral monitoring techniques. Lastly, while the eDNA method showed high sensitivity, further investigation is needed to clarify the relationship between eDNA concentration and actual infection status, as well as the stability and degradation of eDNA under various environmental conditions. Nevertheless, this study offers essential baseline data that will contribute to the development of effective virus monitoring and early warning systems for aquaculture farms.