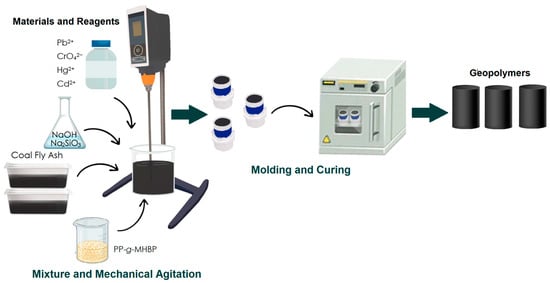
3.2.1. Compressive Strength Test Analysis
The results in
Table 4 indicate a gradual increase in the compressive strength of each specimen group as the curing time progressed. At 7 days, the strength was lower, reaching its maximum value at 28 days. This pattern reflects the ongoing geopolymerization process [
40,
41].
At 7 and 14 days, specimens with HM ions showed increased PE of compressive strength compared to GP1, likely owing to differences in cation charge densities, sizes, and hydration patterns, which influence geopolymerization [
42]. However, at 28 days, the compressive strength decreased in all HM-containing specimens compared to that of GP1, suggesting a threshold for HM ion incorporation without structural compromise. HM ions may interfere with geopolymerization by reducing alkalinity, increasing viscosity, and hindering the dissolution of silica and alumina, thereby weakening the geopolymer network [
4,
43].
Specimens containing CrO
42− exhibited distinct behavior. At 28 days, specimens with 3.0 wt% chromium exhibited only a slight reduction in compressive strength compared to other HM-containing specimens. This trend aligns with previous findings [
9,
42], which suggest that CrO
42− promotes the polymerization of [SiO
4]
4− and [AlO
4]
5− units, facilitating their incorporation into the three-dimensional amorphous gel structure. Oxyanionic species such as CrO
42− influence geopolymerization by promoting condensation reactions in highly alkaline environments. While these species may not form covalent bonds with the aluminosilicate framework, their presence in Na
+-rich media enhances the dissolution of aluminosilicate precursors, leading to accelerated formation of the amorphous gel structure [
44]. This mechanism likely explains the relatively high compressive strength observed in specimens with 3.0 wt% CrO
42−.
The compressive strength values for GP1-Hg-3.0 and GP1-Pb-3.0 at 28 d were similar, within the limits of their standard deviations, indicating no significant difference between these specimens.
At 28 days, other factors, such as the particle size disparity between CFA and PP-g-MHBP, also appeared to contribute to the observed strength reduction. Nonuniform mixing likely caused changes in the packing density, porosity formation, and particle interactions, which, in turn, reduced the mechanical strength.
Another plausible explanation for the compressive strength reduction observed at 28 days is the alkaline hydrolysis of ester linkages in PP-g-MHBP in the highly alkaline environment of the GPs. This mechanism can degrade the polymeric structure and weaken its reinforcing effect. Although not directly investigated in this study, this warrants further investigation.
According to the US EPA, a material cured for 28 days should exhibit a compressive strength of at least 0.35 MPa to be considered stable for the safe disposal of hazardous waste [
35]. Regardless of the incorporated HM ions, all specimens in this study demonstrated compressive strengths well above this threshold, with GP1 exhibiting the highest value. Furthermore, 69% of specimens exceeded 10 MPa.
3.2.2. XRD Analysis
The XRD patterns of the CFA sample, GP1, and the GPs containing HM ions at 1.0 wt% and 3.0 wt% after 28 d of curing are presented in
Figure 4, with the identified phases listed in
Table S2. Two types of peaks were observed: (a) residual peaks from the original CFA and (b) secondary peaks formed during geopolymerization.
The spectra of the CFA and GP1 specimens are shown in
Figure 4a. The CFA profile exhibited a broad hump at 2θ, between 15 and 40°, which was attributed to the amorphous or vitreous fraction. The characteristic crystalline phases of CFA include low quartz, mullite, magnesioferrite, and hematite. In contrast, the GP1 profile revealed changes in the vitreous hump, reflecting the structural reorganization of the amorphous fraction owing to Na-ion incorporation and hydration state changes in the Si-Al lattice [
5]. A reduction in the intensity of crystalline peaks associated with CFA, such as quartz and mullite, suggests partial dissolution of these phases in the alkaline solution, consistent with previous reports, indicating their nonreactive nature [
45,
46]. These phases likely act as aggregates or inert fillers within the GP matrix [
19,
45]. Additionally, thermonatrite (Na
2CO
3·H
2O), a secondary phase commonly formed during geopolymerization, was detected. As is extensively documented, this phase results from the reaction between free NaOH and atmospheric CO
2 [
5,
47].
The XRD patterns of GP1 and the GP specimens containing HM ions at 1.0 wt% and 3.0 wt% are shown in
Figure 4b and
Figure 4c, respectively. The spectra were similar across the specimens and were dominated by crystalline phases from CFA, such as low quartz and mullite, which appeared unaffected by geopolymerization. However, a decrease in the quartz peak intensity compared with CFA indicates the formation of a geopolymeric gel [
48]. Changes in the amorphous halo (2θ range: 15–40°) in the GP specimens, particularly a shift to the right, suggest the formation of a geopolymeric gel with medium- and long-range disorder [
5,
49]. This gel comprises Si and Al atoms in a tetrahedral arrangement, forming a three-dimensional network balanced by Na
+ cations from the activator solution [
12].
The addition of HM ions slightly altered the intensity of the amorphous halo; however, these changes were challenging to quantify. The slight changes observed in the amorphous halo intensity in the XRD patterns with the addition of HM ions were interpreted qualitatively because of the limitations of the current data. The decrease in halo size in the HM-containing specimens compared to GP1 suggests reduced amorphous gel formation, potentially explaining the lower compressive strength observed in these specimens. Future studies employing advanced techniques such as PONKCS QXRD could quantitatively evaluate these changes and validate these observations.
No crystalline phases containing Hg or Cd were detected in GPs containing these metals. If these cations had formed a crystalline phase, significant changes in the diffraction peaks would have been observed because of the differences in ionic radii and the consequent deformation of the crystalline unit cells. Because this did not occur, it is reasonable to assume that crystalline phases containing Hg or Cd were not formed during the geopolymerization process or that most phases incorporating these cations were amorphous, making them undetectable by XRD. These findings are consistent with those of previous research [
4,
8]. However, a calomel phase (HgCl
2) was detected in the sample with 3.0 wt% Hg
2+. The presence of this phase in the XRD diffractogram can be attributed to various factors, including the intentional introduction of HgCl
2 during the synthesis of the GP specimens, incomplete dissolution of HgCl
2 during specimen preparation, and the possibility of excess unreacted HgCl
2 being freely present in the specimens, which is related to the limited retention of Hg
2+ within the geopolymeric structure, as detailed in
Section 3.2.6.
In specimens containing Cr
6+, the presence of Cr-doped mullite was confirmed, consistent with previous studies [
8]. This result suggests that CrO
42− was reduced to Cr
3+ during geopolymerization, which was potentially mediated by Fe
2O
3 and SO
3 in the CFA. The proposed reduction mechanism is provided in
Section 3.2.7 (CrO
42−). This reduction in oxidation state may have significant implications for the toxicity and compressive strength of these specimens [
50], as detailed in
Section 3.2.1.
In specimens with Pb
2+, cerussite (PbCO
3) was identified and formed during geopolymerization via chemical carbonation. The low solubility of PbCO
3 likely contributes to Pb stabilization by reducing Pb leaching [
6]. However, carbonation may adversely affect the mechanical strength by reducing the pH and activation rates, thereby introducing structural discontinuities that act as stress concentrators. This effect likely explains the reduced compressive strength of the lead-containing specimens compared to that of GP1 [
51,
52].
No specific crystalline phases associated with PP-g-MHBP were detected in the XRD patterns of the GP specimens, likely because of the low concentration of the polymer. This suggests that PP-g-MHBP has a limited effect on the dissolution of CFA components, such as quartz or mullite. Although the carboxylate groups in PP-g-MHBP were hypothesized to enhance the HM ion retention, the XRD data did not reveal the stabilization or capture of these ions in the crystalline phase. These interactions may have occurred primarily in the amorphous phase. The results of the other characterization techniques discussed below support this observation.
3.2.3. ATR-FTIR Spectroscopy Analysis
During geopolymerization, sodium aluminosilicate hydrate (N-A-S-H) is the primary product of the low-calcium GPs [
3,
53]. This gel is predominantly amorphous with a limited tendency to crystallize [
9], making its detection by XRD challenging owing to the diffuse signals typical of amorphous materials.
Figure 5 presents the IR spectra of the CFA sample, control specimen (GP1), and specimens with varying Pb
2+/CrO
42−/Hg
2+/Cd
2+ contents. The assignment of the characteristic IR bands based on previous studies is summarized in
Table 5.
The ATR-FTIR spectrum of CFA (
Figure 5 and
Table 5) revealed nine distinct minima in transmittance. The minimum at 3736 cm
−1 is attributed to the stretching vibrations of -OH groups from water molecules. The minima located at 1649 and 1512 cm
−1 are assigned to the bending vibrations of H
2O molecules, suggesting the presence of adsorbed water [
54]. The broad and intense minimum at approximately 1055 cm
−1 is associated with the asymmetric stretching vibrations of Si-O-T bonds (T = Si or Al), which are mainly present in quartz, mullite, and the vitreous phase [
55]. Additionally, the presence of quartz in the CFA was confirmed by the minima located at 689 and 459 cm
−1 [
54]. Finally, the presence of mullite was indicated by minima at 773, 594, and 552 cm
−1 [
56,
57].
Table 5.
Assignment of characteristic minima in the ATR-FTIR spectra of the CFA sample and GP specimens.
Table 5.
Assignment of characteristic minima in the ATR-FTIR spectra of the CFA sample and GP specimens.
| Wavenumber (cm−1) | Assignment | References |
|---|
| 3850–3362 | Vibrational stretching of hydroxyl groups (-OH) | [54,56] |
| 2992–2978 | Asymmetric stretching vibrations of the C-H bonds in the CH3 groups | [58] |
| 1694–1639 | Bending vibration of H–O–H in adsorbed water molecules | [20,56] |
| 1543–1377 | Asymmetric stretching of O–C–O in
including the carbonation of the amorphous gel, and asymmetric stretching of -N-O in | [7,56] |
| 1055 | Asymmetric stretching of Si-O-T (T = tetrahedral Si or Al) in the vitreous phase (may partially overlap with mullite and quartz) | [55,57] |
| 988–978 | Asymmetric stretching of Si-O-T in geopolymeric gel, possibly with higher Al concentration | [20,34] |
| 773–766 | Asymmetric stretching of Si-O-Si and stretching of Al-O in sixfold coordination | [8,55] |
| 689–675 | Symmetric stretching of Si-O-T | [43,56] |
| 596–584 | Bending of Si-O-T in mullite or mullite-like structures | [11] |
| 554–548 | Symmetric stretching of Al-O-Si in mullite or mullite-like structures | [43,55] |
| 501–490 | Bending of T-O in TO4 tetrahedra | [56,59] |
| 461–417 | Bending of O-Si-O and Si-O-Si in Si-rich glass or quartz | [55,60] |
The effect of geopolymerization on the ATR-FTIR minima of the CFA sample in the absence of HM ions was elucidated by comparing the spectra of GP1 and CFA (
Figure 5 and
Table 5). The main vibrational signal, centered at approximately 1055 cm
−1 and attributed to the asymmetric stretching vibrations of Si-O-T bonds, shifts to a lower wavenumber (988 cm
−1) after this process. This shift could be explained by two factors. First, the formation of an amorphous alkali aluminosilicate, N-A-S-H gel, resulting from the alkali activation of CFA, indicates the presence of tetrahedral aluminum in the gel structure [
60,
61]. Second, the shift may result from an increase in the proportion of Si or Al sites with non-bridging oxygens (NBOs) such as Si-O- and Al-O-, which are caused by the interaction of these sites with alkali ions (Na
+) from the activating solution [
20,
34].
Compared to the CFA spectrum, the ATR-FTIR spectrum of GP1 reveals the appearance of a new minimum in transmittance located at approximately 1441 cm
−1. This minimum is attributed to the asymmetric stretching vibrations of the O-C-O bonds, which are characteristic of thermonatrite (Na
2CO
3·H
2O) [
5,
7]. The formation of thermonatrite is attributed to the reaction of CO
2 from the air with the residual NaOH in the GP matrix, generating carbonates and water as byproducts [
60]. This observation is further corroborated by the XRD analysis, which confirms the presence of the thermonatrite phase in GP1.
Other differences between the CFA and GP1 spectra are evident in the signals located between 773 and 400 cm
−1, which are associated with quartz and mullite in the CFA. Specifically, the minima around 459 and 773 cm
−1, assigned to the T-O (T = Si or Al in tetrahedral coordination) bending modes and the Al-O bending of the octahedra aluminum (AlO
6), respectively, showed a decrease in their intensities after the geopolymerization process, particularly in the signal at 773 cm
−1. This suggests a partial breakdown of the original structure of the aluminum octahedra present in mullite [
59].
The effect of incorporating Pb
2+, CrO
42−, Hg
2+, and Cd
2+ in the geopolymerization process is evident when comparing the spectrum of GP1 with those of specimens containing HM ions (
Figure 5 and
Table 5). The spectra of HM-containing specimens display minima between 3389 and 3362 cm
−1, which is attributed to O-H stretching vibrations, which are absent in the CFA sample and GP1. These bands were associated with the formation of solvated cations, M(H
2O)ₙ (M = Na
+, K
+), as part of the porous GP structure [
20]. Furthermore, an increase in the intensity of the minima between 1645 and 1639 cm
−1, linked to H-O-H bending vibrations, indicates a higher presence of non-structural Si-O-H and Al-O-H bonds hydrogen-bonded to water molecules (Si-OH···H
2O and Al-OH···H
2O) in specimens with higher HM ion content [
20,
41].
These changes suggest a possible interaction between the HM ions and hydroxyl ions (OH
−) in the activating solution. During geopolymerization, HM ions compete for OH
− or disrupt the reaction, thereby affecting the dissolution of CFA. This resulted in specimens with greater quantities of OH
−, H
2O, and M(H
2O)
n complexes [
7,
8]. Excessive HM ion addition reduces the alkali available for geopolymerization, hindering the polymerization of silicate and aluminosilicate units and leading to reduced geopolymer formation. This reduction explains the decrease in compressive strength observed in specimens with HM ions compared to GP1, particularly as the HM ion content increases, except in specimens with CrO
42−.
In specimens with HM ions, weak minima are observed in the range of 2992 to 2978 cm
−1, corresponding to the asymmetric stretching vibrations of C-H bonds in CH
3 groups, which is attributed to the presence of PP-
g-MHBP and organic matter in the CFA [
58]. Minima in the 1385–1377 cm
−1 range indicate asymmetric stretching vibrations of O-C-O bonds, suggesting the presence of carbonate salts and the carbonation of the amorphous gel [
5,
60]. Furthermore, the amplitude of these minima suggests their origin as a combination of Na
2CO
3, NaHCO
3, and HM carbonates. The XRD results support this observation, confirming the presence of thermonatrite in GP1 and cerussite in specimens GP1-Pb-1.0 and GP1-Pb-3.0. However, in other GP specimens, carbonate phases were not clearly identified by XRD (
Figure 4), likely because of their low concentrations or reduced crystallinity [
54].
Other researchers have attributed the minimum at 1385 cm
−1 to the asymmetric stretching vibrations of N-O groups from residual nitrate groups, which is plausible given that lead and cadmium were incorporated as nitrates. Additionally, the higher Cd
2+ content in the GP specimens corresponds to an increased minimum intensity, suggesting its relationship with NaNO
3 formation during the geopolymerization process [
7,
56], or that a portion of the initial Cd(NO
3)
2·4H
2O may not have fully integrated into the matrix.
The incorporation of HM ions in the geopolymerization process also influences the minimum transmittance. Initially centered at 988 cm
−1 in the IR spectrum of GP1, this minimum shifted to lower wavenumbers (986–978 cm
−1) in specimens with HM ions. This shift indicates the formation of a geopolymeric gel and an increase in the Si-O
−Na
+ and Al-O
−Na
+ bonds, which are capable of coordinating HM cations [
20,
62]. Cations such as Pb
2+, Hg
2+, and Cd
2+, due to their ionic radii being similar to Na
+, may partially replace Na
+ cations at Si and Al sites with non-bridging oxygens (NBOs) during the polycondensation process, facilitating their immobilization.
Zhang et al. reported that a greater shift in the main minimum correlates with a stronger impact of HM ions on a geopolymeric network [
18]. This aligns with the findings of the present study (
Figure 6). An increase in the HM ion content in GP specimens led to a more pronounced shift in the transmittance minimum and a corresponding decrease in compressive strength at 28 days, particularly in specimens containing Pb
2+, Hg
2+, and Cd
2+. These results suggest that the substitution of Na
+ cations by HM cations at the Si-O
− and Al-O
− sites reduces the polymerization of the gel network and shifts Si-O-T vibrations to lower energies.
Although the overall charge balance is maintained during this substitution, the larger ionic radii and higher polarizability of the HM cations introduce local distortions in the gel network, reducing its connectivity and mechanical strength. Additionally, the release of Na+ ions during substitution may act as structural modifiers within the three-dimensional network, further contributing to the observed reduction in compressive strength.
In GP specimens containing CrO42−, the opposite trend is observed. As the content of CrO42− ions in the GP matrix increased, a smaller shift in the main transmittance minimum and a lower loss of compressive strength were observed. This behavior is attributed to the reduction of CrO42− ions in the presence of OH− during the geopolymerization process, resulting in the formation of Cr3+ cations. These Cr3+ cations may substitute for Al3+ in the primary geopolymer structure and/or in the mullite phase present in CFA, as suggested by the chromium-doped mullite phase detected by XRD in specimens with CrO42−. The increased presence of this compound likely helped maintain the strength, explaining the lower loss of compressive strength observed at 28 d in the GP1-Cr-3.0 specimen.
Finally, another effect of HM ions on the GP specimens is observed in the minima recorded between 775 and 685 cm
−1, which indicates the presence of amorphous aluminosilicate with ring- and cage-like structures. The cavities within these structures act as adsorption and retention sites for the HM cations [
21].
Figure 5 shows that the intensity of the minima at 775 and 677 cm
−1 in specimen GP1 increased with the incorporation of HM ions into the GP matrix. This finding suggests that although HM ions reduce the polymerization of the gel, they favor the formation of ring- and cage-like structures over a fully polymerized network.
Therefore, based on the ATR-FTIR findings, the role of PP-g-MHBP in facilitating the capture or stabilization of HM ions remains unclear. Further studies are required to clarify this impact. Insights into the elemental distribution and potential interactions of these ions within the GP matrix were provided by the SEM-EDX analysis discussed in the subsequent subsection.
3.2.4. SEM-EDX Analysis
The micrographs and EDX spectra of the fractured surfaces of GPs with 1.0 wt% and 3.0 wt% Pb
2+, CrO
42−, Hg
2+, and Cd
2+ ions after 28 d of curing are shown in
Figure 7a–h and
Figures S1–S4. The SEM-EDX analysis provided the general elemental composition of the specimens, representing an average of the tested regions. Although these results offer valuable insights into the overall GP matrix, localized variations in elemental distribution, particularly near cracks or unreacted particles, may not be fully captured. This limitation should be considered when interpreting the data, especially in areas where microstructural defects, such as cracks, may influence the elemental distribution and mechanical properties.
Two types of CFA particles were identified in all the SEM images: partially reactive and unreacted. Geopolymeric gels are formed through the dissolution and activation of vitreous CFA spheres using an alkaline activator. Unreacted CFA particles are predominantly surrounded by or partially embedded within these geopolymeric gels, which consolidate and encapsulate some of the partially reactive CFA particles.
The increased number of unreacted CFA particles observed in the specimens can be attributed to two factors. First, the presence of HM ions affects the dissolution rates of Si and Al in the GP matrix. Specifically, the introduction of Pb(NO3)2, K2CrO4, HgCl2, and Cd(NO3)2·4H2O was correlated with a higher quantity of unreacted CFA particles, thereby reducing the formation of geopolymeric gels. This suggests that HM ions slowed down the reaction process of GPs by inhibiting the dissolution of the aluminum and silicon phases. Secondly, the rapid initial water loss during furnace heating is likely to hinder geopolymerization, further increasing the amount of unreacted CFA particles. The influence of these factors on the dissolution and aggressive water loss during heating likely explains the higher presence of unreacted CFA particles as the HM ion content increases.
Micropores were observed on the surface of the GPs, likely due to air bubbles trapped during the preparation process. Additionally, concave voids from partially split cenospheres (hollow particles from CFA) were identified, with some voids partially filled with the geopolymerization products. Numerous cracks were also visible in the micrographs (
Figure 7), as the width of the cracks increased as the weight percentage of the incorporated Pb
2+, Hg
2+, and Cd
2+ ions increased. This behavior correlates with the decrease in compressive strength observed in specimens with a higher wt% of these ions.
In contrast, the specimens containing chromium exhibited fewer cracks as the CrO
42− wt% increased. This trend aligns with the ATR-FTIR analysis, which shows a smaller shift in the main transmittance minimum and a reduced loss of compressive strength as CrO
42− wt% increases. The reduction of CrO
42− ions to Cr
3+ during geopolymerization, as indicated by ATR-FTIR and XRD analyses, suggests that Cr
3+ cations may substitute for Al
3+ in the geopolymer structure, potentially stabilizing the matrix and reducing crack formation. When the Cd
2+ content reached 3.0 wt% (
Figure 7h), a crack with an approximate width of 2 µm was observed, which was possibly caused by the rupture of the GP specimen’s structure during strength testing.
Table 6 presents the atomic concentrations (%) of the elements in the GP specimens after 28 days of curing, as determined by general SEM-EDX analysis. The primary elements detected were O, C, Na, Mg, Al, Si, K, Ca, Ti, and Fe; O, Na, Al, and Si were identified as crucial for the geopolymerization process. Furthermore, as the concentration of HM ions (Cd
2+, Hg
2+, CrO
42−, and Pb
2+) increased, their corresponding contents in the specimens also increased. Atomic ratios such as Si/Al, (Na + K + Ca)/Al, Ca/(Na + K), Na/Al, and Ca/Si were calculated and are summarized in
Table 7.
According to Zhao et al. [
63], geopolymerization products include geopolymeric gels containing calcium, such as calcium-alumino-silicate-hydrate (C-A-S-H) and sodium-alumino-silicate-hydrate (N-A-S-H), when (Na + K + Ca)/Al ≤ 0.95. These gels coexist as calcium silicate hydrate (C-S-H) and N-A-S-H gels when (Na + K + Ca)/Al > 0.95 [
63]. In this study, despite the low Ca content in the CFA, SEM-EDX analysis detected Ca in all GP specimens, with the (Na + K + Ca)/Al ratio exceeding 0.95 in every case, indicating the presence of both N-A-S-H and C-S-H gels in the GP matrix. Given the higher reactivity of Ca
2+ compared to Na
+ and K
+, Ca
2+ ions are more likely to react with SiO
42− ions or Si-O-Si bonds, promoting the formation of C-S-H gels [
63].
Additionally, the calculated Ca/(Na + K) ratios, ranging from 0.01 to 0.03, and Ca/Si ratios, from 0.01 to 0.02, suggest that calcium is present in smaller amounts relative to the combined concentrations of sodium, potassium, and silicon. This further supports the hypothesis that N-A-S-H gels are more prevalent than C-S-H gels in the matrix.
The SEM/EDX analysis and atomic ratios in
Table 7 reveal a clear relationship between the chemical composition of the specimens and their mechanical behavior, as observed in the micrographs and compressive strength results. In the Cd
2+ specimens, the Si/Al ratio significantly decreased from 3.69 to 2.15 as the cadmium concentration increased. This correlates with a higher occurrence of cracks and compressive strength loss ranging from 11 to 36%. The (Na + K + Ca)/Al ratio also decreased, suggesting that Cd
2+ negatively affected the structural cohesion of the GP matrix, reducing the formation of stabilizing gels.
Similarly, in the Pb2+ specimens, the Si/Al ratio remained consistent at approximately 1.85 but exhibited more cracks and a compressive strength loss of 14–42% as the lead content increased. Although the aluminosilicate network remained stable, Pb2+ interfered with the formation of N-A-S-H and C-S-H gels, thereby weakening the matrix. In contrast, the Hg2+ specimens showed an increase in the Si/Al ratio but suffered significant compressive strength losses at high mercury weight percentages, suggesting that mercury induces structural defects despite enhanced silica polymerization.
Conversely, the CrO42− specimens demonstrated greater structural stability, with an improved Si/Al ratio and fewer cracks, resulting in minimal compressive strength loss. This suggests that Cr stabilizes the matrix by forming Cr3+, contributing to improved cohesion and mechanical strength compared to other HM ions.
Although the initial hypothesis proposed that PP-g-MHBP could enhance the retention of HM ions within the GP matrix through its carboxylate groups, SEM-EDX analysis did not provide clear evidence to support this. While HM ions were detected in the specimens, the increased structural defects, such as cracks, observed at higher weight percentages of Pb2+, Cd2+, and Hg2+, suggest that PP-g-MHBP does not significantly improve the distribution or stabilization of these ions. In contrast, CrO42− specimens exhibited fewer cracks and improved matrix cohesion, likely due to the stabilizing effect of Cr3+ ions formed during geopolymerization. Furthermore, no clear improvement in mechanical properties was observed with the inclusion of PP-g-MHBP. These results suggest that HM ions disrupt matrix cohesion, and the role of PP-g-MHBP in HM ion stabilization remains inconclusive based on these findings.
These findings highlight the varying effects of HM ions on compressive strength and structural integrity and provide valuable insights into how specific ions can either stabilize or degrade the GP matrix.
3.2.5. XPS Analysis
XPS analysis was used to evaluate the surface composition of the GP1 and GP specimens synthesized with 1 wt% Pb
2+, CrO
42−, Hg
2+, and Cd
2+. The wide-scan XPS spectra (
Figure 8) reveal characteristic peaks of oxygen (O 1s, O KLL), carbon (C 1s), silicon (Si 2p, Si 2s), aluminum (Al 2p, Al 2s), and sodium (Na 1s, Na 2s), which is consistent with the expected composition of the GP matrix.
The detection of the Na KLL peak in the XPS spectra confirmed the presence of Na on the GP matrix surface. The Na 1s peak, associated with Na
+ ions, suggests their incorporation into the geopolymeric structures. Although these peaks do not directly reveal the role of sodium in stabilizing the geopolymeric structure or immobilizing HMs, they align with the ATR-FTIR results. The band at ~988 cm
−1 indicates the formation of N-A-S-H gels stabilized by Na
+ ions, as reported in previous studies [
7,
12]. Thus, XPS provides the surface-level elemental composition, while ATR-FTIR complements it by highlighting the role of sodium in N-A-S-H gel stabilization.
The combined ATR-FTIR and XPS results indicated that Pb2+, CrO42−, Hg2+, and Cd2+ ions significantly influenced the N-A-S-H gel structure. The ATR-FTIR band shift to lower wavenumbers suggests an increase in NBO sites, with Na+ stabilizing the negative charges of tetrahedral [AlO4]−. This aligns with the Na 1s peak shifts in XPS; a decrease in binding energy was observed for specimens with Pb2+, CrO42−, and Hg2+ compared to GP1, suggesting interactions between these metals and sodium. Conversely, an increase in the Na 1s binding energy in the Cd2+ specimen suggests distinct interactions, likely due to differences in ionic size or charge distribution affecting the Cd coordination in the geopolymeric network. These interactions modify the chemical environment of sodium, potentially redistributing or altering its coordination with the tetrahedra [SiO4] and [AlO4]−. This redistribution affects the N-A-S-H gel polymerization and contributes to the reduced compressive strength of the specimens with HMs. The interaction of metal ions with the GP matrix disrupts the charge balance and gel structure, explaining both spectral shifts and mechanical property degradation.
The binding energy values for Na 1s and other elements are detailed in
Tables S3–S7, which present the chemical speciation and atomic concentrations of the elements in the GP specimens. These data support the observations presented in this section.
Chlorine (Cl 2p) was detected in all specimens except GP1-Pb-1.0, likely originating from trace amounts in the raw materials, such as CFA, as confirmed by XRF analysis, or from synthetic compounds, such as HgCl2 in GP1-Hg-1.0. The Cl 2p signal corresponds to Cl− species, indicating that chlorine is primarily present as chloride ions on the GP matrix surface. In contrast, SEM-EDX analysis did not detect chlorine in any specimen (GP1 and those with 1 wt% HM ions), suggesting that chlorine was concentrated in the outermost layers, which is consistent with the higher surface sensitivity of XPS. This supports the hypothesis that chloride ions are likely to be adsorbed on the GP matrix surface, originating from trace components in the raw materials or synthetic reagents.
The high-resolution XPS spectra (
Figure 9) revealed peaks for Si 2s (
154 eV), Al 2s (
119 eV), and Na 2s (
64 eV), confirming the presence of these elements on the GP matrix surface. The peaks for Si 2p (
103 eV) and Al 2p (
74 eV) present in all the specimens are typically associated with Si-O and Al
3+ species in the [SiO
4]
4− and [AlO
4]
5− tetrahedral networks, respectively. The shifts to lower binding energies in the Si 2p and Al 2p peaks for specimens with HMs, compared to GP1, suggest interactions with the GP matrix, likely increasing the electron density around the Si and Al atoms and distorting the Si-O and Al-O bonds. The XRD and ATR-FTIR results corroborate this, showing geopolymeric gels with medium- and long-range disorder and a shift of transmittance minima to lower wavenumbers, indicating reduced polymerization. These structural changes align with the decreased compressive strength in HM-containing specimens, particularly owing to reduced amorphous gel formation and the presence of cage-like structures in ATR-FTIR. Further studies are needed to fully understand the extent of these distortions, as the current results provide indirect yet significant evidence of HM-induced structural changes.
In the GP1-Pb-1.0 specimen, two distinct peaks at 139.06 eV and 144.03 eV were observed, corresponding to Pb 4f7/2 and Pb 4f5/2, respectively. These peaks confirm the presence of Pb2+ in the GP matrix, with a 5 eV separation characteristic of spin–orbit coupling, indicating that lead is predominantly in the +2 oxidation state (Pb2+). As expected, no Pb peaks were detected in GP1, as no Pb was added. The Pb2+ peaks in GP1-Pb-1.0 confirmed the successful incorporation of Pb into the GP matrix and its detectability on the surface by XPS. Additionally, the Pb 4f peaks indicate that Pb2+ retained its oxidation state from that of the original Pb(NO3)2, demonstrating its stability during geopolymerization.
In contrast, no clearly defined peaks corresponding to Hg, Cr, or Cd were observed in the XPS spectra (
Figure 8 and
Figure 9). This can be attributed to their low concentrations on the GP surface, resulting in weak and noisy signals that hinder quantification. These signals overlapped with more intense Si and Al peaks. Although auxiliary signals have been used to accurately identify metals, weak signals and peak overlaps remain significant challenges. Another hypothesis is that these metals are predominantly distributed within the GP bulk, beyond the XPS detection range (
5–10 nm). The SEM-EDX analysis (
Table 6) partially supports this, detecting small concentrations of Cr, Hg, and Cd, suggesting that these elements are present but not concentrated in the outermost layers.
The data in
Tables S3–S7 reveal changes in oxygen speciation due to the incorporation of HM ions compared to GP1. In GP1-Pb-1.0, the binding energies for (C=O)-OH species are 532.68 and 537.70 eV, showing decreases of 0.51 eV and 0.53 eV compared to GP1 (533.19 and 538.23 eV). These shifts suggest that Pb
2+ alters the chemical environment of oxygen in the GP matrix, likely through the formation of PbCO
3, as confirmed by the XRD analysis.
In GP1-Hg-1.0, binding energies of 532.88, 533.58, and 537.90 eV were observed, compared to GP1. The new binding energy at 533.58 eV, absent in GP1, suggests an interaction of Hg2+ with the oxygen chemical environment, possibly linked to amorphous phases or surface composition changes not detected by XRD. A decrease of 0.31 eV at 532.88 eV and 0.33 eV at 537.90 eV indicates a redistribution of electronic density on the material’s surface. These changes may correlate with the cracks observed in the SEM micrographs and the reduction in the compressive strength of the Hg2+-containing specimens.
For GP1-Cr-1.0, the oxygen binding energies at 533.21, 531.81, and 537.77 eV were detected. Compared to GP1, a decrease of 0.46 eV at 537.77 eV suggests a redistribution of electronic density around oxygen atoms, which is likely due to chromium incorporation into the Si-O and Al-O networks on the matrix surface. This aligns with the detection of Cr-doped mullite, indicating that Cr is structurally integrated into the GP matrix.
In GP1-Cd-1.0, the oxygen binding energies were observed at 533.09, 531.65, and 537.49 eV. The new binding energy at 531.65 eV, absent in GP1, indicates changes in the chemical environment owing to interactions with Cd2+. Additionally, decreases of 0.10 eV at 533.09 eV and 0.05 eV at 537.49 eV suggest local alterations in the oxygen environment, which are likely caused by Cd2+ interactions on the GP matrix surface.
The alterations in oxygen speciation across the specimens highlighted the significant impact of HM ions on the chemical environment of oxygen within the GP matrix. The shifts in binding energies, particularly for the (C=O)-OH species, suggest that HM ion incorporation redistributes the electronic density on the surface, potentially affecting the stability of the GP network.
In addition to oxygen,
Tables S3–S7 also reveal the chemical speciation of carbon on the GP specimen surface, identifying carbonyl (C=O) and carboxyl (O=C-O) groups. These groups may have originated from the carboxyl (-COOH) functional groups in PP-
g-MHBP and carbonaceous compounds or carbonates in the CFA. Combined data from XPS, ATR-FTIR, and XRD provided insights into the interactions between PP-
g-MHBP, GP matrix, and HM ions.
ATR-FTIR spectra displayed weak minima between 2992 and 2978 cm−1, corresponding to the asymmetric stretching vibrations of C-H bonds in CH3 groups, which suggests the presence of organic components from PP-g-MHBP and residual organic matter in the CFA. Minima between 1543 and 1377 cm−1, attributed to the asymmetric stretching vibrations of O-C-O bonds, indicated the presence of carbonate salts, such as Na2CO3, NaHCO3, and HM carbonates.
The XRD results supported these observations by identifying Na2CO3·H2O in the GP1 specimen and PbCO3 in the Pb2+ -containing specimens. In specimens with Cd2+ and Hg2+, carbonate phases were not clearly identified, likely due to the low concentrations or low crystallinity of the carbonates, which is consistent with the lack of a significant increase in O=C-O species in the XPS analysis.
ATR-FTIR results revealed N-O groups, which is consistent with the presence of nitrates. This was corroborated by the XPS data, which identified Cd(NO3)2 species in the GP1-Cd-1.0 specimen, whereas no nitrate species were observed in GP1-Pb-1.0. The presence of nitrates in GP1-Cd-1.0 suggests that some Cd(NO3)2 used during synthesis did not fully decompose.
Finally, the absence of significant interactions between the carboxylate groups in PP-g-MHBP and the HM ions, as indicated by the XPS results, suggests that other mechanisms, such as carbonate formation, may play a more prominent role in the immobilization of these metals within the GP matrix.
3.2.6. TCLP Leaching Test Analysis
A TCLP test was performed to assess the environmental risks of HM leaching from CFA and GP samples. This test replicates the acidic conditions typically present in landfill environments where contaminants may leach from materials, thereby providing an estimate of their environmental impact. The concentrations of Pb, Cr, Cd, and Hg in the leachate were compared with the maximum allowable limits set by the US EPA. If the metal concentrations remained below these thresholds, the immobilization mechanism was deemed effective, indicating the successful retention of metals within the GP matrix under the test conditions [
17].
Table S8 summarizes the concentrations of HMs in the TCLP extract for the CFA and GP specimens with and without exogenous metals. The results showed that the HM concentrations in both CFA and GP1 specimens were below the EPA allowable limits, suggesting their suitability for environmental applications. However, varying degrees of leaching were observed in all GP specimens, indicating that the release of toxic metals into the leachate was influenced by the nature and concentration of the metals. This behavior aligns with prior research [
5,
18], which showed that metal leaching is closely tied to specific interactions between the metal species and the GP matrix.
Table S8 also presents the percentage of immobilization (PI) for each HM calculated using the following equation [
11]:
where is the HM concentration in the initial TCLP extract, assuming all HMs in the GP specimen are potentially leachable, and is the measured HM concentration in the leachate (mg/L). This calculation quantifies the geopolymer’s ability to retain HMs, with higher PI values indicating more effective immobilization within the matrix. However, a high PI does not necessarily ensure long-term immobilization under environmental stressors such as pH changes, temperature variations, or prolonged exposure to external agents.
Figure 10 illustrates the relationship between HM ion content (1.0–3.0 wt%), HM concentrations in the TCLP extract, PI values, and the compressive strength of GP specimens after 28 days.
Regarding Pb
2+ (
Figure 10a), the Pb concentration in the leachate increases with rising Pb
2+ content in the GP matrix, ranging from 1.57 mg/L at 1 wt% to 5.31 mg/L at 3 wt%, the latter exceeding the EPA limit of 5.0 mg/L. This trend suggests that while the GP matrix effectively immobilized lower Pb
2+ concentrations, its PI decreased slightly, from 99.66% to 99.65%, as the Pb
2+ content increased. Although this difference is minimal, the higher leaching concentration of 3 wt% highlights the reduced capacity of the GP matrix to retain Pb
2+ at elevated levels. Thus, a high PI value does not necessarily guarantee effective immobilization of Pb
2+, particularly at higher concentrations. Additionally, the compressive strength decreased significantly from 12.96 MPa at 1 wt% to 8.63 MPa at 3 wt%, suggesting that elevated Pb
2+ concentrations negatively affected the mechanical integrity of the GP specimens.
For CrO
42− (
Figure 10b), the Cr concentration in the leachate increases significantly from 42.5 mg/L at 1 wt% to 193 mg/L at 3 wt%, far exceeding the EPA limit of 5.0 mg/L. This substantial leaching indicates that the GP matrix is less effective in immobilizing CrO
42− ions than Pb
2+. The PI content decreased from 91.64% at 1 wt% to 87.49% at 3 wt%, highlighting the limited capacity of the GP matrix to retain CrO
42− at higher concentrations. Despite the high PI values, the observed leaching suggests that CrO
42− is not effectively immobilized within the matrix. Interestingly, the compressive strength increases from 11.02 MPa at 1 wt% to 14.43 MPa at 3 wt%, indicating that CrO
42− may enhance the mechanical strength of the GP specimens, even though its leaching behavior remains unfavorable.
For Hg
2+ (
Figure 10c), the Hg concentration in the leachate increases from 5.94 mg/L at 1 wt% to 26.0 mg/L at 3 wt%, both far exceeding the EPA limit of 0.20 mg/L. This indicates that the GP matrix was ineffective in immobilizing Hg
2+, with substantial leaching occurring even at low concentrations. The PI decreased slightly, from 98.84% at 1 wt% to 98.29% at 3 wt%, remaining relatively high despite significant Hg release. This discrepancy suggests that a high PI does not necessarily correlate with effective metal containment, particularly under unfavorable conditions. The compressive strength decreases from 11.38 MPa at 1 wt% to 8.21 MPa at 3 wt%, following a similar trend observed with Pb
2+, where higher metal ion concentrations compromise the structural integrity of the GP matrix.
For Cd
2+ (
Figure 10d), the Cd concentration in the TCLP extract remains below the EPA limit of 1.0 mg/L across all tested concentrations, ranging from 0.240 mg/L at 1 wt% to 0.63 mg/L at 3 wt%. This indicates that the GP matrix was highly effective in immobilizing Cd
2+, as confirmed by the high PI values ranging from 99.60% at 1 wt% to 99. 95% at 3 wt%. Although these high PI values suggest excellent immobilization, further studies are needed to confirm the long-term stability of Cd
2+ under varying environmental conditions. However, the compressive strength decreases with increasing Cd
2+ content, from 13.31 MPa at 1 wt% to 9.66 MPa at 3 wt%. Despite the GP matrix demonstrating excellent immobilization efficiency for Cd
2+, the reduction in compressive strength indicated compromised mechanical properties at higher Cd
2+ concentrations.
XPS analysis revealed Cl on the surface of all specimens except GP1-Pb-1.0, whereas SEM-EDX detected Cl only in GP1-Hg-3.0. This suggests that Cl was primarily concentrated in the outermost layers of the GP matrix. Although chloride ions were adsorbed on the surface, their impact on the leaching behavior, particularly under acidic conditions simulated by the TCLP test, remains unclear. Further studies are necessary to assess the potential role of surface chloride ions in environmental risk, particularly in specimens containing Hg2+, where a higher chlorine content may affect HM mobility.
This study focuses on the short-term immobilization effects of heavy metals within a geopolymeric matrix. Long-term evaluations are essential for understanding metal release under various environmental conditions. Factors such as pH, temperature, and leachate composition significantly influence stability, as has been highlighted in previous studies [
64].
3.2.7. Immobilization Mechanisms
Pb2+
The results of the characterization techniques and tests suggest that Pb
2+ immobilization in the GP matrix occurs through interrelated mechanisms. XRD analysis confirmed the formation of PbCO
3 in the lead-containing specimens, showing that a fraction of Pb
2+ was immobilized through precipitation as PbCO
3, which is an effective mechanism under alkaline conditions owing to its low solubility. However, the TCLP test revealed increased Pb
2+ leaching under acidic conditions (pH 2.88 ± 0.05), with concentrations rising from 1.57 mg/L at 1 wt% to 5.31 mg/L at 3 wt%. This suggests that carbonate phases such as PbCO
3 are less stable in acidic environments. Despite this, the GP matrix achieved an immobilization percentage above 99.6%, significantly reducing lead release under simulated environmental conditions, which is consistent with observations reported in the literature [
17,
22].
SEM-EDX and XPS analyses corroborated these findings and confirmed the immobilization of Pb2+ in the GP matrix. SEM-EDX detected Pb on the surface, which was likely distributed in phases, such as PbCO3. At the same time, XPS confirmed the presence of Pb in its +2 oxidation state, as indicated by the characteristic peaks of Pb 4f7/2 and Pb 4f5/2, reflecting the chemical stability of Pb ions within the matrix. These results suggest that Pb2+ immobilization occurred through a combination of physical encapsulation and precipitation. Although the N-A-S-H gel played a significant role in retaining Pb2+, the compressive strength results indicated that the Pb2+ addition did not enhance the mechanical stability of the matrix and, at higher concentrations (3.0 wt%), may even weaken it.
The interaction of Pb
2+ with the N-A-S-H gel was clearly evidenced by the ATR-FTIR and XPS results. ATR-FTIR revealed a shift to lower wavenumbers in Pb
2+-containing samples, indicating an increase in NBOs within the aluminosilicate network. This increase suggests that structural modifications in the gel were caused by the incorporation of Pb
2+. Additionally, the XPS results showed a decrease in the binding energy of Na 1s, pointing to an ion exchange mechanism where Pb
2+ ions replace cations such as Na
+ or K
+ within the aluminosilicate network. This ion exchange mechanism is widely reported in the literature as a key process for immobilizing heavy metals, including Pb
2+, in GP matrices [
4].
The changes in the binding energies of Si 2p and Al 2p detected by XPS suggest modifications in the coordination of the Si and Al atoms. This suggests that Pb
2+ not only participates in cation exchange but may also influence the local structure of the N-A-S-H gel, potentially forming new covalent bonds with SiO
4. Reports suggest that Pb
2+, with its partial charge and ionic character resembling Si
4+, could substitute Si
4+ in the geopolymeric structure, particularly at SiO
4 sites. Additionally, the Pb-O bond, which has a lower ionic character (
45%) than Al-O or Cd-O (
55%), is more covalent and stronger, reinforcing Pb
2+ immobilization within the matrix [
17]. However, Pb
2+ incorporation may disrupt gel polymerization, thereby increasing the proportion of less-condensed units. This disruption could explain the reduction in mechanical stability observed in the compressive strength results.
CrO42−
The highly alkaline conditions provided by the activator (14 M NaOH solution and Na
2SiO
3), combined with the presence of Fe
2O
3 in the CFA (identified through XRF and XRD), may have partially favored the reduction of CrO
42− during geopolymerization. Although the literature does not directly document Fe
2O
3 from CFA as a reducing agent in alkaline media, intermediate reactions can generate transient Fe
2+ species that are known electron donors. For example, Fe
2O
3 may undergo partial reduction according to the following reaction:
The electrons required for this process may originate from reductive species, such as sulfites (SO
32−), formed through the hydrolysis of SO
3 present in the CFA. Sulfites are well-documented reductants capable of directly reducing Cr(VI).
In this context, the highly alkaline environment coupled with Fe
2O
3 and other reductive species in CFA likely facilitated the partial reduction of CrO
42− to Cr
3+. Once reduced, Cr
3+, introduced as K
2CrO
4, becomes stable under alkaline conditions and can be structurally incorporated into a geopolymeric network. This stabilization occurred primarily through sorption, ion exchange, and isomorphic substitution of Al
3+ in mullite, as confirmed by the detection of chromium-doped mullite in the XRD patterns [
10,
21].
In this study, the resulting Cr
3+ appeared to have been incorporated into the GP structure, particularly in the chromium-doped mullite phase, as evidenced primarily by the XRD results. Previous research has indicated that Cr
3+ can be incorporated into mullite during geopolymerization, likely by isomorphically substituting Al
3+ in the crystalline structure. This substitution occurs without distorting the crystalline lattice, which is supported by the ionic size and charge similarity between Cr
3+ and Al
3+ [
8].
Although XPS did not detect distinct Cr3+ peaks, possibly owing to its low surface concentration in the GP1-Cr-1.0 specimen, it revealed a redistribution of electronic density around the oxygen atoms. This, combined with the XRD evidence of chromium-doped mullite, supports the hypothesis that Cr3+ was structurally integrated into the geopolymer network, likely contributing to its stabilization. However, the evidence presented is indirect, albeit consistent, and complementary, as no single technique has provided definitive confirmation of Cr3+ incorporation. Future atomic-scale analyses, such as Transmission Electron Microscopy (TEM) and X-ray Absorption Spectroscopy (XAS), are recommended to validate this hypothesis. Additionally, complementary electrochemical and spectroscopic techniques could provide further insights into the redox mechanisms underlying the reduction of CrO42− to Cr3+.
XPS analysis revealed a redistribution of electronic density around the oxygen atoms, indicated by a 0.46 eV decrease in the 537.77 eV peak compared to that of GP1. This suggests a possible alteration in the electronic distribution, attributed to the presence of Cr3+ within the Si-O and Al-O bond networks. Combined with the XRD evidence of chromium-doped mullite, these findings support the hypothesis that Cr3+ was structurally integrated into the geopolymer network rather than being merely surface-bound.
ATR-FTIR analysis indicated the presence of the N-A-S-H gel, with a shift in the bands corresponding to Si-O-T (T = Si or Al) vibrations, suggesting structural alterations in the gel, possibly due to Cr3+ interactions with the amorphous geopolymer network. This spectral shift may reflect adjustments in the bonding environment caused by cations such as Cr3+. Additionally, compared to GP1, the decrease in the binding energy of the Si 2p and Al 2p peaks in GP1-Cr-1.0 suggests that the presence of chromium affects the electronic density around these atoms, distorting the Si-O and Al-O bond networks. This distortion could be linked to a reduction in the degree of polymerization within the matrix.
The TCLP test revealed significant Cr leaching, exceeding the limits established by the US EPA, indicating that not all Cr6+ ions were effectively reduced or immobilized. This leaching may be attributed to the amorphous phases or adsorption sites that are less stable under acidic conditions. In addition to isomorphic substitution, less stable or incomplete immobilization mechanisms such as surface sorption, physical adsorption, or entrapment in the amorphous or porous phases of the matrix may contribute to this behavior. This, combined with the electronic redistribution observed in the XPS analysis, suggests that certain immobilization mechanisms were not fully effective under the acidic conditions of the test.
Hg2+
The mechanism of Hg
2+ immobilization was inferred from the results of previously described characterization techniques and tests, which provided complementary insights into the possible mechanisms of Hg
2+ retention within the matrix. A previous study suggests that Hg
2+ can replace cations such as Na
+ and Ca
2+ to balance the negative charge of [AlO
4]
− tetrahedra, forming structures like -Si-O-Al(Hg)- or -Si-O-Al(Hg)-O-Si- within a three-dimensional GP network [
65]. Cation replacement by Hg
2+, commonly referred to as ionic exchange, has been proposed as a key mechanism for Hg
2+ immobilization in GP matrices.
In GP1-Hg-3.0, the XRD analysis revealed the presence of HgCl2, corresponding to the original compound used in the synthesis. This suggests that at 3 wt% Hg2+, HgCl2 was not fully incorporated into the GP matrix, likely because of matrix oversaturation and its limited solubility in water (~6 g per 100 mL at room temperature). Under highly alkaline conditions, Hg2+ may have precipitated as hydroxides, which were even less soluble and stabilized within the matrix over time. These findings highlight the limited capacity of the matrix to immobilize Hg2+ at higher concentrations, leaving residual crystalline HgCl2 unincorporated into the amorphous N-A-S-H geopolymer gel network.
ATR-FTIR analysis of GP1-Hg-3.0 revealed transmittance minima in the range of 3389 to 3387 cm
−1, corresponding to the vibrational stretching of -OH groups, and at 980 cm
−1, which is associated with the asymmetric stretching of Si-O-T bonds (T represents Si or Al in tetrahedral coordination). These minima were absent in GP1, indicating that the incorporation of Hg
2+ promoted additional interactions with hydroxyl groups and altered the amorphous structure of the GP matrix. This aligns with previous studies suggesting that Hg
2+ precipitates as mercury hydroxide (Hg(OH)
2) in alkaline environments, forming stable species encapsulated within the matrix [
65]. At higher Hg
2+ concentrations, such as in GP1-Hg-3.0, these structural alterations may contribute to the reduced stability of the geopolymeric network.
In contrast, the XRD analysis showed no evidence of crystalline HgCl2, indicating a more effective incorporation of Hg2+ into the amorphous structure of the geopolymeric gel at lower concentrations. Additionally, the ATR-FTIR spectrum of GP1-Hg-1.0 showed a transmittance minimum shifted to 986 cm−1 for the asymmetric stretching of Si-O-T bonds, compared to 988 cm−1 in GP1. This shift to lower wavenumbers suggests that, at lower concentrations, Hg2+ interacts more effectively with the N-A-S-H gel network, promoting its retention within the amorphous structure and supporting the hypothesis of more stable Hg2+ immobilization in the GP matrix at low concentrations.
XPS analysis of GP1-Hg-1.0, compared to GP1, revealed a new binding energy at 532.88 eV for the oxygen species (C=O)-OH, suggesting a change in the chemical environment of oxygen, possibly due to the interaction of Hg2+ with oxygen atoms in the matrix. This, combined with shifts to lower binding energies in the Al 2p and Si 2p peaks, indicates redistribution of the electron density within the GP matrix. Such redistribution is likely caused by the incorporation of Hg2+ at the oxygen sites in the SiO4 and [AlO4]− tetrahedra, locally altering the structure of the N-A-S-H gel network.
Additionally, an increase in Al3+ and Si-O concentrations was observed in GP1-Hg-1.0, indicating that Hg2+ may promote localized structural rearrangement within the geopolymeric network, increasing the exposure and availability of oxygen sites in the [SiO4] and [AlO4]− tetrahedra. This adjustment in elemental concentrations suggests that Hg2+ is chemically immobilized within the matrix through interactions with NBOs, facilitating both physical and chemical retention within the amorphous structure.
SEM-EDX confirmed the presence of Hg on the matrix surface, suggesting that Hg2+ was effectively integrated into the amorphous structure. However, at higher mercury concentrations, particularly in GP1-Hg-3.0, more cracks and microdefects appeared in the GP structure. These structural defects correlated with the higher mercury leaching observed in the TCLP test and the reduction in compressive strength.
The TCLP test revealed that even at low concentrations (1 wt% of Hg
2+), mercury is highly leachable under acidic conditions (pH 2.88 ± 0.05), reaching leachate concentrations that exceed US EPA regulatory limits, consistent with previous reports [
12]. The GP1-Hg-1.0 sample exhibited a PI of 99.06%, indicating that a significant fraction of Hg
2+ was retained in the GP matrix. However, at higher Hg
2+ concentrations (GP1-Hg-3.0), the leachate concentration increased to 26.0 mg/L, accompanied by a reduction in the PI to 98.29%. This behavior suggests matrix oversaturation with Hg
2+, which compromises its retention capacity under acidic conditions.
Oversaturation was also associated with a significant decrease in compressive strength, from 11.38 MPa in GP1-Hg-1.0 to 8.21 MPa in GP1-Hg-3.0 at 28 days of curing. Higher Hg2+ concentrations weaken the mechanical integrity of the GP matrix, promoting the formation of cracks and microdefects, which enhance Hg2+ leaching. Additionally, the presence of chlorine on the surface can influence Hg2+ mobility, increasing the leaching risk under adverse environmental conditions. These findings highlight the need for further studies to fully understand the role of surface chlorine in HM mobility within the GPs.
Cd2+
The results from various characterization techniques, combined with compressive strength and TCLP tests, indicate that Cd
2+ in the GP specimens is immobilized through a combination of physical encapsulation and chemical stabilization, which is consistent with findings reported in the literature [
17,
18]. SEM-EDX analysis revealed extremely low atomic concentrations of Cd
2+ on the surfaces of the GP1-Cd-1.0 and GP1-Cd-3.0 specimens (0.00% and 0.01%, respectively), suggesting that Cd
2+ was primarily distributed within the bulk of the GP. This limited surface exposure likely reduced its availability for leaching under acidic conditions. These findings are consistent with the TCLP results, which showed Cd concentrations in the leachate to be below the US EPA limit (1.0 mg/L), confirming the effective immobilization of Cd
2+ within the GP matrix.
XPS analysis revealed that Cd
2+ is present on the surface primarily as Cd(NO
3)
2, with atomic concentrations of 0.025% and 0.017% in GP1-Cd-1.0 at binding energies of 405.71 eV and 412.58 eV, respectively. This finding is corroborated by ATR-FTIR results for GP1-Cd-1.0 and GP1-Cd-3.0, which showed a minimum transmittance in the 1385 cm
−1 region, attributed to the asymmetric -N-O vibration in NO
3− [
7]. These observations suggest the persistence of nitrate groups in the surface layers of GP specimens. The combination of these techniques indicates that part of the initial Cd(NO
3)
2·4H
2O may not have been fully integrated into the matrix, thus remaining in a potentially more leachable form.
The minimum transmittance observed in the 1385–1377 cm
−1 region for specimens with Cd
2+ may also be attributed to the formation of cadmium carbonates [
5,
56], suggesting that Cd
2+ is partially immobilized in the GP matrix as carbonate. Additionally, the transmittance minima at 3389 cm
−1 for GP1-Cd-1.0 and 3387 cm
−1 for GP1-Cd-3.0, along with the minimum at 1645 cm
−1, whose intensity increases with the cadmium weight percentage, indicate the possible formation of Cd(OH)
2. This suggests that Cd
2+ immobilization may also occur via hydroxide precipitation, as reported in previous studies [
18,
66]. The presence of these hydroxides and carbonates in the amorphous or low-crystallinity phases, corroborated by the absence of cadmium crystalline phases in the XRD analyses, likely reduced the mobility of Cd
2+ and its susceptibility to leaching under acidic conditions, as observed in the TCLP test. These findings are consistent with prior research, which also reported the absence of crystalline phases containing Cd [
7].
In the ATR-FTIR spectra, a shift to lower wavenumbers was also observed in the main transmittance minimum, located at 988 cm
−1 for GP1, 982 cm
−1 for GP1-Cd-1.0, and 980 cm
−1 for GP1-Cd-3.0. According to the literature, this shift may be attributed to two possible sub-mechanisms of Cd
2+ immobilization involving covalent bonding [
20]. The first suggests the direct incorporation of Cd
2+ into the interconnected Si and Al networks of GP. However, based on Goldschmidt’s rules, it is unlikely that Cd
2+ would replace Al
3+ or Si
4+ in the aluminosilicate structure due to differences in charge, ionic size, and bonding type. The lower charge of Cd
2+ compared to Al
3+ and Si
4+ makes its direct substitution into these sites challenging without compromising structural stability. Additionally, the Cd
2+ bond type, which has a lower ionic character and reduced stability in a network requiring strong covalent bonds, further limits its compatibility with the Si
4+ positions [
17].
The second sub-mechanism of Cd2+ immobilization involves coordination with NBOs, resulting in the release of Na+ ions within the GP matrix. This excess Na+ may alter the structure by introducing additional ions that, when occupying interstitial sites, reduce the cohesion of the aluminosilicate network and increase porosity. XPS analyses supported this interpretation, revealing a decrease in the binding energies of species assigned to Si-O and Al3+ in GP1-Cd-1.0 compared to GP1, suggesting a weakening of the network and the creation of new NBOs in response to Cd2+ incorporation. Furthermore, the increase in Na+ binding energy indicates redistribution of this ion within the GP environment, likely occupying interstitial sites or positions less integrated into the structural network. These structural effects collectively weaken the matrix, potentially explaining the reduction in compressive strength observed in the Cd2+-containing specimens after 28 days.
XPS results for Al
3+, Si-O, and Na
+ species suggest that Cd
2+ may act as a charge-balancing cation, a mechanism previously reported in the literature [
4]. In GPs, the substitution of Si
4+ by Al
3+ in the aluminosilicate structure generates a negative charge that is typically balanced by monovalent ions such as Na
+. However, Cd
2+, as a divalent ion, may also fulfill this role, although to a lesser extent owing to its higher charge and larger ionic radius. Despite this, the role of Cd
2+ as a charge-balancing cation likely represents a more stable and less disruptive electrostatic interaction than its coordination with NBOs. Based on the compressive strength results, the predominant mechanism of Cd
2+ immobilization appears to be its coordination with the NBOs, as discussed previously.