1. Introduction
For an extended period, meat has been regarded as a key component of the human diet due to its complete profile of essential amino acids and the high digestibility of its protein [
1,
2]. Since 1961, global meat consumption has nearly doubled, increasing from approximately 23 kg per person per year to around 43 kg by 2021 [
3]. This significant increase can be attributed to population growth, urbanization, and evolving dietary preferences, particularly in developing countries where rising incomes have led to increased meat consumption [
4]. In contrast, while developed countries remain substantial consumers, they have experienced a more moderate increase, reflecting changes in dietary patterns [
5,
6]. A key factor driving this shift is the growing concern about the adverse effects of excessive meat consumption on both health and the environment.
Reducing meat consumption in the diet is associated with numerous health benefits, contributing to overall well-being and reducing the risk of chronic diseases [
7]. Many research findings indicate that a decrease in meat intake, particularly red and processed varieties, is correlated with a reduced risk of cardiovascular disease, type 2 diabetes, and certain cancers, especially colon cancer [
8,
9,
10]. There is evidence that decreasing meat consumption can lead to significant environmental benefits, including lower greenhouse gas emissions, less water consumption, and less land degradation. Studies suggest that even a slight reduction in meat intake can lead to a significant decrease in resource consumption and environmental impact [
11,
12,
13].
As consumers become increasingly aware of their dietary choices, many are deciding to reduce their meat consumption or seek alternative protein sources that align with their health and ethical considerations [
14,
15]. Consequently, the growing demand for meat analogs has led to the emergence of innovative food technologies aimed at meeting consumer preferences, not only in terms of nutritional value but also with regard to sensory characteristics [
16,
17].
Despite the increasing popularity of meat analogs, major challenges remain in optimizing production processes. A primary obstacle in developing meat analogs is achieving a texture that closely resembles the fibrous, elastic, and juicy characteristics of an animal muscle [
18]. Traditionally, high-moisture extrusion has been employed, applying heat and shear forces to a protein mixture, aligning the proteins into a fibrous structure that partially mimics meat. However, while extrusion can produce meat-like textures, it has notable limitations. This process requires high energy and precise temperature control to prevent protein degradation [
19]. Furthermore, the extrusion process can impart undesirable off-flavors and odors to the final product, which may be unappealing to consumers looking for a close replication of meat’s similar profile [
20]. To overcome these challenges, researchers are exploring alternative methods to enhance the physical and functional qualities of meat analogs. A promising approach involves the incorporation of hydrocolloids and enzymatic preparations into the meat analog formulations [
21].
Incorporation of hydrocolloids, such as alginate and methylcellulose, into meat analogs presents a promising alternative to extrusion, offering flexibility in texture formation. Hydrocolloids are well-known for their ability to form gels and modify the texture and moisture retention properties of food products [
22]. When combined with proteins, hydrocolloids can create hydrocolloid-protein biocomposites that mimic the microstructural characteristics of meat [
23]. When a solution containing a protein isolate and sodium alginate is injected into a calcium chloride solution, gelation occurs due to calcium ions cross-linking alginate molecules. This results in the formation of fibers that closely resemble the structure of meat fibers while maintaining a high protein content [
24]. Proteins participate in creating the external appearance, color, juiciness, and texture of food due to its functional properties including gelation, solubility, water holding capacity, emulsification, and nutrition value [
25].
Transglutaminase is an enzyme that occurs naturally in various organisms. Initially, this enzyme was obtained from guinea pig liver, fish tissue, and plant tissue. It was soon discovered in microorganisms grown in appropriate media synthesize extracellular transglutaminase, which is then isolated using methods such as ultrafiltration, evaporative concentration, or salting out with inorganic salts. Unlike animal transglutaminase, microbial transglutaminase is calcium-independent, has a smaller molecular weight, and is much cheaper to produce [
26]. The Food and Drug Administration approved the use of microbial transglutaminase as a “Generally Recognized as Safe—GRAS” for food processing in 1998. Microbial transglutaminase is considered safe, nontoxic, nonallergenic, nonimmunogenic, and non-pathogenic for public health. Additionally, there is no requirement to list it among the products’ ingredients [
27].
Microbial transglutaminase plays a key role in the formation of meat analogs by catalyzing protein cross-linking, specifically through transamidation. This reaction involves the formation of covalent bonds between proteins by linking amino acid residues of glutamine and lysine. This process can occur both intramolecularly (within a single protein molecule) and intermolecularly (between different protein molecules), leading to the formation of protein polymers [
28]. By facilitating this process, microbial transglutaminase strengthens the protein matrix, imparting elasticity and firmness to the product [
29]. This combination not only stabilizes the fibrous structure formed during ionic gelation but also provides a texture that provides a better imitation of the chewiness and elasticity of meat fibers. Enzymatic treatment improves texture attributes such as hardness and cohesiveness, which are critical for consumer acceptance of meat analogs [
30].
The advantages of using protein–hydrocolloid composites cross-linked by microbial transglutaminase lie in the lower energy input compared to the extrusion process and the fact that they do not rely on high temperatures, preserving the integrity and nutritional quality of the proteins. Furthermore, these composites allow for improved control over mechanical strength by adjusting microbial transglutaminase concentration and incubation time [
31]. Consequently, the main objective of this study was to propose a protein texturization process using microbial transglutaminase to produce a meat-like texture and structure by investigating the effects of varying concentrations of microbial transglutaminase and different incubation times on the rheological properties of composite materials, along with an assessment of their quality parameters.
2. Materials and Methods
2.1. Materials
Egg protein albumin was obtained from Ovopol Sp. z o.o., while acid casein (85.7% proteins) was produced by Polsero Sp. z o. o. (Sokołów Podlaski, Poland). Microbial transglutaminase, derived from the culture of Streptoverticillium spp.—MTG, ACTIVA WM (99% maltodextrine and 1% microbial transglutaminase; activity of ≈96 U/g) was purchased from Ajinomoto Foods Europe SAS (Paris, France). Sodium alginate FD 125 and methylcellulose (Methocel™) were procured from Dupont GRINSTED® (Grindsted, Denmark and Dow Wolff Cellulosics Gmbh (Bomlitz, Germany), respectively. The commercially available wheat fiber WF 300 and pea fiber EF 150 were sourced from Vitacel, Rettenmaier (Warsaw, Poland). Anhydrous calcium chloride was purchased from the company P.P.H. “STANLAB” s.j. (Lublin, Poland) and canola oil was obtained from a local supermarket (Tesco, Wrocław, Poland). Flavor additives and colorants used included cochineal red E124 and brown HT E155 (Food Colours Perczak S.J., Piotrkow Trybunalski, Poland), sodium chloride, ground black pepper, herbal pepper, ground white pepper (McCormick Polska S.A., Stefanowo, Poland), a chicken-vegetable bouillon cube (Prymat Sp. z o.o., Jastrzebie Zdroj, Poland), and sausage flavoring (ŻUK—POL Sp. Z o. o., Wroclaw, Poland).
2.2. Methods
2.2.1. Samples Preparation
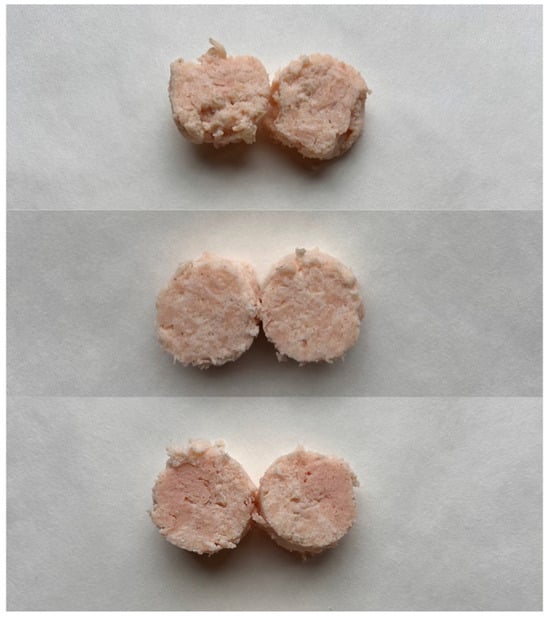
The process of preparing chicken meat sausage analog began with the formulation of the protein solution in a 250 mL beaker. The combination of 68.5% water and 7.5% canola oil was heated to 70 °C using a water bath (TW 12 series, JULABO GmbH, Seelbach, Germany) and stirred with a R50 CAT mechanical stirrer (Ballrechten-Dottingen, Germany) fitted with a propeller blade at 350 rpm. Additional ingredients were added gradually, increasing the stirring speed to 600 rpm. First, 8% acid casein was stirred for 10 min, then 1.9% sodium alginate was stirred for 15 min, followed by 0.3% methyl cellulose for 3 min, and finally 2.7% flavoring and 2% coloring was added.
The development of protein–hydrocolloidal fibers involved the addition of anhydrous calcium chloride (0.6%) to a protein solution, which was kept at 70 °C. A mechanical stirrer and a flat-ended spatula were used to mix the solution at 600 rpm, preventing fiber clumping and ensuring a consistent blend. Afterward, the mixture was allowed to cool to room temperature, at which point the remaining ingredients were added manually. Albumin was incorporated at a concentration of 6%, followed by the addition of fibers—WF300 and EF150—each at 1%, and MTG at varying concentrations of 0, 0.5, and 1 (in powder preparate) depending on the specific variant presented in
Table 1.
The blend was subsequently sealed in 23 mm diameter cellulose casings (Lommel Belgium) using a stuffing machine and secured with butcher’s string. The samples were then incubated in an incubator (UNE 400, Memmert, Schwabach, Germany) at 55 °C for 90 or 180 min or left unincubated. After this incubation period, the samples were pasteurized to reach an internal temperature of 70 °C, cooled to ambient temperature in cold water, stored in the refrigerator for 24 h and then re-warmed to room temperature prior to analysis (
Figure 1).
2.2.2. pH Determination
The measurement of acidity was performed for each sample after 24 h of refrigeration, using a pH meter (inoLab, Weilheim, Germany). Prior to analysis, the device was calibrated using buffer solutions (pH 4 and 7). The pH value was obtained by immersing a calomel electrode in the prepared homogenates.
2.2.3. Protein Content Determination
The total nitrogen content was determined using the Kjeldahl method with the Kjeltec 2300 Analyzer (FOSS, Hillerod, Denmark), in accordance with the standard PN-75/A-04018 [
32]. This method involves the conversion of organic nitrogen compounds into (NH
4)
2SO
4 through the application of concentrated H
2SO
4 in the presence of a catalyst K
2SO
4 and CuSO
4 × 5H
2O. Subsequently, the solution was alkalized, distilled, and titrated with a 0.1 N HCl solution until a pink-violet color was achieved. The protein content was calculated using a universal nitrogen conversion factor of 6.25.
2.2.4. Determination of Dry Matter
This determination was performed based on the Polish Standard PN-ISO 1442:2000 [
33], utilizing a thermal drying method. Samples of 3 g of the ground material were prepared and weighed with an accuracy of 0.001 g. The samples were dried at a temperature of 103 ± 2 °C until a constant weight was achieved. The samples were then cooled in a desiccator until they reached room temperature. Then, the samples were weighed again with an accuracy of 0.001 g. The difference in mass of the samples before and after drying represented the dry matter content. Calculations were performed using the following formula:
where: DM—dry matter [%]; a—mass of the sample after drying [g]; b—mass of the sample before drying [g].
2.2.5. Determination of Fat Content and Dry Matter of Defatted Mass
The determination was conducted according to the Polish Standard PN-ISO 1444:2000 [
34] using an extraction method with a Soxhlet apparatus. The principle of the method is based on the extraction of a dried sample using petroleum ether. The solvent residue was removed through evaporation and drying, ensuring that the samples reached a constant weight. The difference in mass before and after extraction represented the fat content. To calculate the fat content, the following formula was employed:
where FC—free fat content in the product [%]; M—mass of the filter paper with the fat sample after drying [g]; m—mass of the filter paper after extraction and drying [g]; n—mass of the fat sample before drying [g].
For the calculation of the dry matter of defatted mass, the formula used was
where DMDF—dry matter of defatted mass [%]; DM—dry matter [%]; FC—free fat content in the product [%].
2.2.6. Identification of the Total Water and Fat Leakage, the Water and the Fat Content in the Total Leakage
The determination was performed according to the method established by Lee and Patel [
35]. Samples measuring 15 mm in height were cut from cylindrical blocks with a diameter of 20 mm. Each sample was placed between two pre-weighed Grade 1 (Whatman, Maidston, UK) filter papers, with a precision of 0.001 g, and subsequently compressed to a 40% deformation relative to the initial height of the sample using the Z010 testing device (Zwick Roell, Ulm, Germany). When the specified deformation level was reached, compression was halted, and the sample was kept under load for 60 s. Subsequently, the compressing head was retracted to its original position, and the samples were removed from between the papers, which were then weighed again with a precision of 0.001 g. The weighed papers were dried at a temperature of 103 ± 2 °C until a constant mass was achieved.
Based on the measurements obtained, the total water and fat leakage of the sample (TL), the water content in the total leakage (WL), and the fat content in the total leakage (FL) were calculated. The formula used to determine the total water and fat leakage is as follows:
where TL represents the total water and fat leakage from the sample [%]; mpa denotes the mass of the paper after compression [g]; mpb indicates the mass of the paper before compression [g]; M represents the mass of the sample [g].
For calculating the water content in the total leakage, the formula used is
where WL indicates the water content in the total leakage [%]; mpa is the mass of the paper after compression [g]; mpd is the mass of the paper after drying [g]; M denotes the mass of the sample [g].
To calculate the fat content in the total leakage, the following formula was employed:
where FL denotes the fat content in the total leakage [%]; TL represents the total water and fat leakage from the sample [%]; WL indicates the water content in the total leakage [%].
2.2.7. Thermal Loss Determination
To assess the mass loss resulting from thermal processing, samples were weighed both prior to and after pasteurization. Losses were calculated as the difference in mass of the samples before and after thermal treatment. The results were presented as the efficiency of the process.
2.2.8. Determination of Selected Textural Parameters
The determination of the texture profile and selected viscoelastic parameters was performed using the Z010 testing device (Zwick Roell, Ulm, Germany). Samples were analyzed after being stored for 24 h under refrigeration, following a prior heating to room temperature. Cylindrical samples (15 mm × 23 mm, H × d) were cut from the bars. These samples underwent a 75% deformation test through double compression (TPA) and included a relaxation time of 30 s between compressions. For the TPA test, five rheological parameters were quantified: hardness [N] was defined as the maximum force applied to the sample during initial compression. Springiness [-] indicated the distance traveled by the probe during the second compression cycle. Cohesiveness [-] was the ratio of work undertaken in the second compression to that in the first. Gumminess [N] was calculated as the product of hardness and cohesiveness, while chewiness [N × mm] was determined as the product of gumminess and springiness [
36].
2.3. Statistical Analysis
Statistical analysis of the results was performed using GraphPad Prism software, version 10.3.1 (464) (GraphPad Software, San Diego, CA, USA). A two-way analysis of variance (ANOVA) was applied to process the data, and the significance of differences between groups was evaluated using Tukey’s post hoc test, with the significance level set at 0.05. All evaluations were performed in triplicate unless mentioned otherwise.