1. Introduction
Chinese cabbage (
Brassica rape L. ssp.
Pekinensis) is a significant vegetable in Asia, with its consumption steadily increasing in Western countries [
1,
2]. However, the growth of Chinese cabbage is negatively impacted by abiotic stress, which leads to reductions in both yield and quality, thereby significantly affecting agricultural productivity. Abiotic stress refers to the detrimental effects on the normal growth and development of plants caused by non-biological factors. Common abiotic stressors that plants frequently encounter include drought, radiation, nutrient deficiency, extreme temperatures (both high and low), metal ion toxicity, salinity, and organic pollution, all of which severely compromise the distribution and growth conditions of plants worldwide [
3]. Research indicates that abiotic stresses such as drought, extreme temperatures, and high salinity influence nearly every stage of the Chinese cabbage life cycle, affecting not only the expression of relevant genes but also cellular metabolism and developmental processes [
4]. Under these stress conditions, Chinese cabbage can develop a certain degree of resistance; however, as the intensity of the stress escalates, this self-generated resistance becomes insufficient to cope with more severe abiotic challenges. The consequences of such stress can result in abnormal growth or even mortality of the Chinese cabbage, leading to diminished yields and subsequent market shortages. The capacity of plants to respond to environmental changes is critical for their adaptation and survival. Hence, it is essential to investigate the mechanisms by which plants adapt to varying climatic conditions. Consequently, researching the molecular mechanisms underlying abiotic stress in Chinese cabbage is of great significance for enhancing its yield under adverse conditions. In particular, the identification of abiotic stress-responsive genes (SRGs) [
5] and proteins is vital for fostering the resilience of Chinese cabbage [
6].
In recent years, the advancement of high-throughput technologies has led to the rapid generation of extensive biological data. The availability of complete genome sequences of various plant species has enabled comprehensive research on biomacromolecules, thereby promoting the investigation of abiotic SRGs at the whole-genome level. In addition to the identification of SRGs through transcriptome analysis, gene expression studies serve as an additional method for recognizing these genes [
7,
8,
9,
10,
11,
12,
13]. Specifically, 35 BrHsf genes have been identified in Chinese cabbage, and a thorough analysis has revealed their potential to enhance plant heat tolerance [
10]. Furthermore, comparative genomic analyses have enhanced our comprehension of the evolutionary dynamics of Hsf genes in cabbage. A co-expression network of genes responsive to cold, drought, and salt stress in cabbage has been constructed and analyzed from multiple perspectives, leading to the identification of previously unknown genes associated with abiotic stress tolerance [
4]. Additionally, through the evolutionary analysis of gene families, abiotic SRGs have also been identified within the B-box family in cabbage. The application of advanced analytical methods to high-throughput genomic data—including genes, transcripts, proteins, and metabolites—will undoubtedly optimize data utilization and enhance the accuracy of abiotic SRGs identification [
14].
In the domain of bioinformatics, various machine learning algorithms have been employed to address significant biological challenges. The integration of machine learning into biological research has introduced a novel perspective that contrasts sharply with traditional experimental and simulation methodologies. This approach has demonstrated considerable potential due to its flexibility, accuracy, and robust generalization capabilities when analyzing complex biological systems [
15]. In the early stages of genomic research, gene identification primarily relied on biological experiments, gene sequence analysis, and traditional statistical methods. This phase often involved a considerable amount of manual analysis and experimental validation. Researchers employed basic computational techniques, such as sequence alignment and pattern recognition, to identify genes; however, these methods frequently depended on manual rules and intuitive judgment [
16]. With the development of high-throughput sequencing technologies, genomic data began to grow explosively [
17]. At this point, traditional manual analysis methods and single statistical approaches struggled to handle such vast amounts of data. Therefore, machine learning techniques began to see preliminary applications in gene identification [
18]. Subsequently, the scale and complexity of genomic research have significantly increased, with the intricate relationships among genes, phenotypes, and the environment becoming the focal point of study. At this juncture, the application of machine learning has diversified, particularly in areas such as gene function prediction and gene–phenotype association analysis. In recent years, the use of machine learning has evolved toward a more integrated and multifaceted approach. With the advent of genomic, transcriptomic, epigenomic, and other types of data, machine learning has played a crucial role in synthesizing these diverse data types and conducting multi-level gene characterization analyses [
19]. The widespread application of machine learning in gene identification and characterization is expected to steer future research toward a greater emphasis on model interpretability, thereby providing scientific evidence to support clinical and agricultural decision-making.
Artificial intelligence-driven machine learning techniques have emerged as pivotal tools for data interpretation, particularly in the context of predictive modeling and plant stress responses [
20]. Previous research has successfully predicted the types of stresses to which plants respond by analyzing the expression patterns of plant microRNAs (miRNAs). In this context, intricate non-linear relationships between input variables (miRNA expression) and output variables (plant stress responses) are discerned from training datasets housed in various databases. This enables the identification of whether previously uncharacterized plant miRNAs are responsive to stress conditions [
21]. Additionally, computational models utilizing machine learning have been developed to predict proteins associated with abiotic stress in plants [
22], specifically focusing on the classification of abiotic stress response proteins in crops of the family Poaceae through the application of deep convolutional neural networks [
14]. These investigations have illustrated the efficacy of machine learning methodologies in the identification, classification, and prediction of stress-responsive molecules in plants. However, despite the labor-intensive and time-consuming nature of identifying genes related to abiotic stress through conventional genetic techniques, there remains a lack of dedicated computational models for the identification of abiotic SRGs in Chinese cabbage. Given these considerations, the development of a computational method to predict abiotic SRGs in Chinese cabbage is warranted.
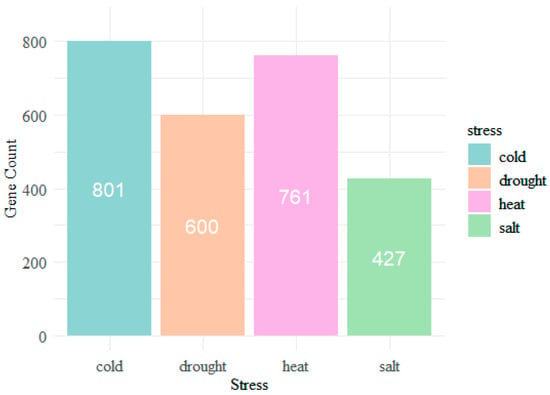
The objective of this study is to develop a machine learning-based computational model to identify the genes associated with cold, heat, drought, and salt stresses in Chinese cabbage. This endeavor aims to uncover novel abiotic SRGs within this species. It is hypothesized that the stress resistance phenotype of Chinese cabbage is influenced by multiple genes or genomic regions (quantitative trait loci, QTLs) and that there exists a correlation between these genes and various abiotic stresses, including cold, heat, drought, and salt. By analyzing the complex relationship between the genotype of Chinese cabbage and these stress resistance phenotypes using machine learning methods, relevant stress resistance genes can be identified. The relationship between genotype and stress resistance phenotype is not merely linear; there may also be complex non-linear or higher-order interactions. These complex relationships may involve interactions between genes, responses of phenotypes to environmental changes, and so on. Machine learning, especially algorithms like random forests (RF), support vector machines (SVM), and deep learning, can capture these complex non-linear patterns, thus effectively mining these stress resistance genes. Focusing on our goal, we collected and organized genes responsive to the four types of stress from related articles, further obtaining the protein sequences formed by the transcription and translation of each gene. We used the protein sequence as an effective high-throughput data format to analyze the specific targets related to abiotic stress, establishing complex linear relationships between the protein-coding sequences of Chinese cabbage genes and the four types of stress.
4. Discussion
Abiotic stress is a major limiting factor affecting the yield and quality of crops, including Chinese cabbage, a highly nutritious and economically important vegetable. Investigating the molecular mechanisms behind abiotic stress responses in Chinese cabbage is essential for developing stress-resistant varieties and enhancing agricultural productivity. The foundation of traditional methods is rooted in experimental techniques that directly observe and verify molecular or physiological mechanisms [
59]. However, these methods frequently encounter challenges such as high costs, low efficiency, and complex data [
60]. With the development of modern technology, an increasing number of methods are beginning to integrate experimental and computational approaches, thereby enhancing the efficiency of discovery [
61]. Compared to machine learning, deep learning models require a large amount of data to train their complex network structures, while small datasets often lack sufficient support for effective model generalization [
20]. Furthermore, traditional algorithms tend to be more efficient in feature engineering and model training, rendering them more suitable for practical applications involving small datasets. Consequently, when working with limited data, it is generally more reasonable to prioritize traditional machine learning methods.
To explore potential solutions, this study employed machine learning models to predict novel genes that may be linked to abiotic stress. Through this approach, we aimed to identify candidate genes that could be involved in stress responses, providing insights into their potential roles in stress tolerance. Compared to previous research [
22,
62,
63], this study achieved an average auROC score of around 0.8, with a maximum score of 0.88. Additionally, this method not only contributes to uncovering the molecular factors associated with abiotic stress in crops like Chinese cabbage but also serves as a valuable tool for guiding the development of stress-resistant varieties and improving crop productivity.
A key challenge in this research is that a single gene can be associated with multiple types of stress, underscoring the complexity of abiotic stress responses [
22]. To address this challenge, we developed four distinct prediction models, each tailored to a specific type of stress. If a multiclassification model were employed to predict a gene’s response to multiple stresses simultaneously, the sum of the probabilities for all four stresses would equal to 1. This approach would result in predicting only the stress with the highest response probability for each gene, thereby failing to account for the possibility of a gene responding to multiple stresses. To overcome this limitation, we constructed separate models for each type of stress.
In this study, various feature construction methods were employed, and the best performing features were selected through comparative analysis to develop a more robust and comprehensive model. To minimize the risk of overfitting, enhance the model’s generalizability, and accelerate training speed, we utilized the SVM-RFE method for feature dimensionality reduction. To further enhance model performance, we explored feature combination strategies, where multiple features were combined to improve overall accuracy. The fundamental value of feature combination lies in its ability to expand the feature space, enabling the model to capture complex patterns within the data more effectively [
64]. This enhancement, in turn, enhances both predictive performance and interpretability. In practical applications, feature combination can be utilized alongside methods such as feature selection to further optimize model performance.
Given that combining various feature construction methods, feature selection, and feature combinations would significantly increase computational complexity, we initially screened six feature construction methods—CKSAAP, DDE, DPC, PAAC, APAAC, and TPC—to evaluate their performance across multiple machine learning models. From this evaluation, CKSAAP and DDE consistently outperformed the other feature sets. Consequently, we focused on these two methods for subsequent feature selection and combination, while excluding the remaining four feature sets, particularly PAAC and APAAC, which showed subpar performance, with average auROC scores around 0.65.
Using auROC as the performance metric, we applied SVM-RFE to select the optimal feature subsets for each construction method. Even though the full feature set maximized the auROC score, we discovered that the selected feature subsets outperformed it, achieving our objective of enhancing model performance with a more compact set of features. Further training with the selected feature sets—CKSAAP, DDE, and the combination of CKSAAP and DDE—across various machine learning models enabled us to identify the most robust and high-performing models. From the perspective of the auROC, CKSAAP consistently outperformed the other feature sets across all four types of stress considered in this study. As a result, CKSAAP was identified as the final feature set. We then finetuned the model’s hyperparameters using this feature set to optimize the model’s performance. This process resulted in the selection of the best model for each type of stress, which served as the final results.
Despite the promising results, several challenges persist in this field. Continued advancements in high-throughput sequencing, data standardization, and algorithm development are essential for overcoming limitations related to data quality and model interpretability. The integration of heterogeneous datasets remains a critical challenge in biological research. The effective fusion of multisource data, such as transcriptomics, proteomics, and metabolomics, presents a promising solution to enhance the predictive power and biological relevance of machine learning models [
65]. The explosive growth of biological data has underscored the need for multimodal integration, as combining diverse data types—such as genomic sequences, protein structures, and metabolic profiles—can provide a more comprehensive understanding of biological processes [
15]. This integration has been shown to significantly improve model performance, leading to more accurate and biologically relevant predictions that better capture the complexity of biological systems.
5. Conclusions
In this study, a novel machine learning-based computational tool was introduced to predict stress-related proteins, marking a significant advancement over traditional methods such as BLAST. This tool demonstrated strong potential in identifying genes associated with cold, heat, drought, and salt stress, providing a solid foundation for further functional studies and crop improvement efforts.
This study identified several novel cold, heat, drought, and salt stress-related genes in the Chinese cabbage genome, many of which have functional support in the literature for other plant species. These findings lay a solid foundation for future experimental validation and functional characterization under abiotic stress conditions. These efforts will contribute to a deeper understanding of stress resistance mechanisms and promote the development of stress-tolerant crops. Furthermore, the associated online prediction server provides a user-friendly platform for researchers, facilitating the translation of computational findings into experimental applications.
In conclusion, this study highlights the transformative potential of machine learning in crop stress research. By enabling the identification and characterization of stress-related genes, it lays the groundwork for developing stress-resistant crop varieties. As climate change and environmental pressures intensify, such innovations will be crucial in ensuring sustainable agricultural production and global food security.