1. Introduction
End-stage renal disease (ESRD), the most advanced stage of chronic kidney disease (CKD), is highly prevalent in the United States: the United States Renal Data System reports over 2219 per million population and 135,972 new cases in 2021 alone. Of these, 83.8% underwent in-center hemodialysis (HD); nonetheless, ESRD patients on HD faced a mortality rate of 191.5 per 1000 person-years, with infections and cardiovascular disease as the most common causes [
1]. ESRD manifests as a duality of immune incompetence driven by uremic toxicity, which leads to increased risk of infection, co-existing with immune activation driven by the accumulation of proinflammatory cytokines, which contributes to the progression of atherosclerotic lesions and vascular disease [
1,
2]. The uremic state in ESRD is known to contribute to the chronic activation of natural killer (NK) cells, monocytes, dendritic cells (DCs), T cells, and B cells and the production of proinflammatory cytokines that predisposes these cells to anergy, apoptosis, and overall premature immunological aging that could be traced to epigenetic remodeling [
3]. As a result, this duality has not only led to higher susceptibility to infection, but also lower response to vaccination: higher vaccination failure rates have been reported for hepatitis B virus, influenza virus,
Clostridium tetani, and
Corynebacterium diphtheriae [
4,
5].
More recently, studies of the COVID-19 BTN162b2 mRNA vaccine in HD patients presented a more optimistic outlook compared to responses to prior vaccines, demonstrating seroconversion rates as high as 96%, though there was still some reduction in SARS-CoV-2 IgG antibodies in HD patients [
6,
7,
8,
9,
10]. The improved immune protection has been attributed to the novel design philosophy of mRNA vaccines, which engages both innate and adaptive components of the immune response through local inflammation at the site of injection as well as replicating key features of natural viral infection, including the production of antigenic proteins by host cells and the induction of type I interferon (IFN) and downstream Th1 polarization, without the same risks that live vaccines would pose to immunocompromised populations [
11]. At the same time, our group previously reported delayed transitioning from the innate immune response to the adaptive immune response in HD patients compared to healthy controls (HCs) over the course of the two-dose BTN162b2 vaccination regimen, likely as a product of chronic immune activation/desensitization in this patient population [
10]. Work by other groups has also demonstrated a decreased innate immune response to SARS-CoV-2 glycoprotein stimulation in HD patients compared to HCs at 4–5 and 8–9 weeks post-vaccination [
12]. Further complicating these matters are conflicting reports of both innate and adaptive immune mechanisms underlying ESRD [
13,
14,
15,
16].
While transcriptomic approaches have tackled this complexity by extracting key differentially expressed genes associated with immune phenotypes, there is growing recognition for examining the regulatory processes that govern gene expression as well. Biological networks offer a holistic characterization of the complex dysregulated interactions among genes and regulators in ESRD [
17]. At the most fundamental level, these networks consist of nodes, which typically represent genes or other biological targets of interest, connected to other nodes by edges, which can represent co-expression or other types of relationships of relevance [
18]. When these relationships can be quantified, e.g., as correlation coefficients, network analysis can aid in pinpointing the most significant relationships from background noise. Therefore, we propose to investigate mechanisms of regulatory dysfunction underlying the delayed immune response to COVID-19 (BNT162b2) mRNA vaccination in HD subjects as compared to healthy controls (HCs) at the (1) transcriptional, (2) epigenetic, and (3) cellular levels by means of network analysis centered around (1) gene module co-expression and (2) gene transcriptional regulation, and combined with (3) immune cell populations computationally deconvoluted from expression data (
Figure S1).
3. Discussion
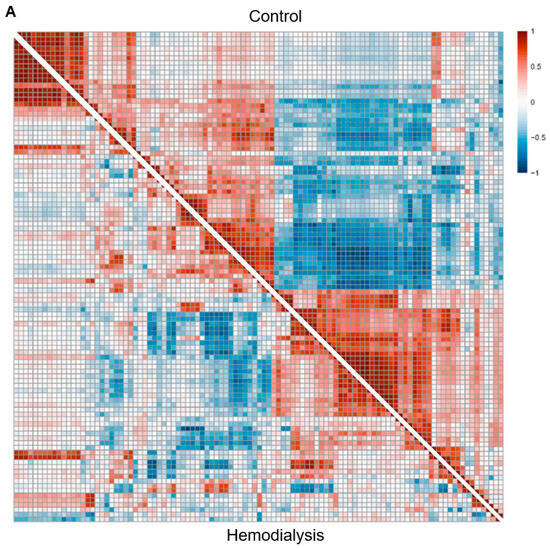
Through BTM co-expression and gene regulatory network analyses, we illustrate how the delayed immune response to COVID-19 mRNA vaccination in the immunocompromised HD population is complicated by the defective coregulation of innate immune signaling. Our BTM co-expression network results demonstrate broadly weakened coupling between different components of the immune system in HD subjects compared to HCs, representing a global desensitization likely driven by chronic inflammation. In particular, BTMs in the DC/APC family exhibited the most weakened positive intrafamily coregulation. These network findings are further contextualized by our cell deconvolution analysis of conventional DC subtypes, in which a significantly higher proportion of cDC2s was noted with a concomitantly lower, albeit non-significant, proportion of cDC1s prior to vaccination in HD subjects compared to HCs. As a result, both the network and deconvolution results are consistent with evidence from the literature showing significantly decreased numbers of DCs in ESRD, which decline further with HD treatment [
40], as well as the impaired maturation of monocytes and DCs and decreased antigen presentation [
41,
42,
43].
The analysis of the BTM co-expression networks additionally showed weakened positive and negative interfamily coregulation between the DC/APC, IFN type I, and Myeloid/Inflamm families. DC dysfunction in ESRD has been proposed to stem from alterations in pattern recognition receptors (PRRs), including both the increased and decreased expression of TLR4 [
13,
44], increased expression of the secreted PRR mannose-binding lectin, and increased expression of major macrophage scavenger receptors SR-A and CD36 [
14]. Interestingly, LI.M146 (MHC-TLR7-TLR8) was another BTM found to exhibit weakened targeting in HD subjects, and features the core enrichment genes TLR7 and TLR8, which have been shown to induce type 1 IFNs in DCs that synergize with the NFkB pathway to activate DCs [
45]. Furthermore, the most significantly weakened edge in each of these interfamily relationships involving the DC/APC family was LI.M43.0 (Myeloid, dendritic cell activation via NFkB (I)), which is also the second most significantly dysregulated BTM from our PANDA network analysis, with core enrichment genes including NFkB pathway mediators such as TNF, NFkB2, and NFkBID. TNF, a proinflammatory cytokine that is upregulated by TLR binding and required for NFkB activation and DC maturation [
46,
47], is also required for both CD8+ T cell activation and apoptosis [
48]. As effector CD8+ T cells possess the potential to differentiate into memory T cells, which can persist for as long as 10 years post-activation, decreased apoptosis may also lead to chronically elevated memory T cell populations [
49]. As a result, its dysregulation of CD8+ T cell apoptosis may account for the chronically elevated CD8+ T cell populations seen in HD, as noted in this study as well as others [
50,
51]. Taken together, these results reinforce evidence of TLR dysfunction, with a mediating role of type 1 IFN and NFkB induction, leading to the impaired maturation and activation of DCs.
In addition to BTMs related to myeloid cells, the most significantly dysregulated BTM from our PANDA analysis was LI.M61.0 (enriched in NK cells (II)), with core enrichment genes comprising activating and tolerogenic receptors on T cells and NK cells. Though deconvoluted NK cell proportions did not significantly differ between HD and HC, there were significant reductions in CD4+ T cells at V1D7 and in NK T cells, which possess receptors characteristic of both NK and T cells [
52] at V2D0 and V2D7 in HD relative to HCs. TGFBR3 and IL2RB are two core enriched genes that play a critical role in the balance of activation and tolerance via Tregs [
24,
27]. The weakened regulation of these genes may, thus, contribute to the disturbed Treg function reported in ESRD [
53,
54] as well as the significantly lower Treg proportions we observed in HD subjects at V1D7. In addition to Tregs, the IL-2 receptor is also involved in the differentiation of anti-inflammatory Th2 cells, which have been reported to be diminished in HD patients and may be reflected in the significantly lower CD4+ proportions observed in our HD subjects at V1D7.
Our regulatory network results from PANDA further identified regulators of cell survival and apoptosis in ESRD. The ESRD literature has demonstrated the accelerated apoptosis of neutrophils [
55] as well as mixed findings of increased B cell apoptosis in one study [
15], in contrast to increased B cell survival factors in another study [
16]. Our results showed the weakened regulation of LI.M94.0 (growth-factor-induced, enriched in nuclear receptor subfamily 4), with differential targeting of three core enriched genes involved in apoptotic signaling (NR4A1, PPP1R15A, and CDKN1A). In conjunction, the deconvolution analysis of B cell subtypes in HD subjects revealed a significant decrease in naïve B cells relative to HCs’ proportions at V1D7, potentially as a result of increased apoptotic activity following vaccine-induced immune stimulation. In contrast, plasmablasts exhibited significantly higher proportions in HD than in HC subjects at V1D0 and V1D7. Immature B cells were shown to be elevated in another study on ESRD patients with HD treatment [
56] and generally persist in this state in response to chronic stimulation [
57]. Nonetheless, both naïve B cells and plasmablasts normalized to HC levels during V2D0 and V2D7 while mature B cell populations remained comparatively similar to HCs throughout the vaccine regimen, suggesting that the earlier observed discrepancies in B cell populations may be a product of delayed upstream signaling. This is consistent with the comparable antibody production and neutralizing function observed in mRNA-vaccinated ESRD patients relative to healthy controls, as previously demonstrated by our group and others [
6,
7,
8,
9,
10]. The mixed findings of B cell apoptosis in ESRD are also likely to be context-dependent: Fernández-Fresnedo et al. cultured peripheral blood cells for four days prior to assessing apoptosis, while Pahl et al. assessed apoptosis on freshly isolated cells.
There is a wealth of evidence demonstrating TLR-induced alterations of the epigenetic landscape, leading to both increased and decreased expression of TLR-induced genes [
58]. For example, in macrophages, LPS signaling through TLR4 alters chromatin accessibility at TLR-responsive inflammatory genes including IL-6 [
59]. In support of a mediating role of type 1 IFN in the TLR dysfunction leading to the impaired maturation and activation of DCs, type I IFN has also been shown to catalyze the methylation of promoters of NF-kB-responsive genes [
60]. Additionally, oxidative stress has been shown to alter DNA methylation profiles, including in peripheral blood. In fact, oxidative damage to a methyl-CpG site in a methyl-binding protein recognition sequence has been shown to substantially reduce the binding affinity of MECP2 [
61]. It is reasonable that, in addition to altering the regulation between immune players, epigenetic mechanisms could independently increase the susceptibility of immune subsets to apoptosis.
Our study is limited by its small sample size due to the selection of patients with samples collected across all time points. However, this is partly mitigated by allowing paired analysis across the time points [
62]. Significant differences in mean age, racial/ethnic background, and past medical history commonly associated with ESRD between the HD and HC groups could additionally influence the differences observed between the two. Specifically, others have demonstrated weaker antibody immune responses in patients over 50 years old versus patients younger than 50 years old to the BNT162b2 vaccine [
63]. While the insights made discernible through our bioinformatics approaches have basis in, and additionally expand on, findings from previous studies, they could be further validated through more direct measurements of the epigenetic and cellular changes in ESRD PBMCs using approaches such as ATAC-seq, DNA methylation profiling, and flow cytometry.
Overall, we elucidated a complex regulatory interplay in ESRD with HD resulting in the simultaneous dampening of immune activation and tolerogenic immune responses that can be appreciated on both a group- and single-subject level. Moreover, by integrating our findings at the epigenetic, transcriptional, and cellular biological levels, we gained a more holistic view of which aspects of the immune system are perturbed and how dysregulation at one level may affect downstream immune cell populations and activity. Our results reinforce prior proposals that TLR dysfunction leads to the impaired maturation and activation of DCs in the HD population. Constitutive stimulation of TLRs may lead to low-grade baseline inflammation, simultaneously resulting in desensitization that impairs the ability of the immune system to mount immunogenic responses. While this may have stymied successful seroconversion by traditional vaccine modalities, mRNA vaccines may still mount robust, if delayed, antibody responses in HD patients comparable to their immunocompetent counterparts by stimulating both the innate and adaptive branches of the immune response. mRNA vaccines thus present a promising avenue to promote the immune-responsive vaccination of ESRD and HD patients against COVID-19 and other high-risk infections.
4. Materials and Methods
4.1. Cohort Recruitment and Sample Acquisition
The study was approved by the University of Illinois at Chicago IRB (#2018-1038) Ethics Review Committee. All research was performed in accordance with relevant regulations, and informed consent was obtained from all participants. Our recruited cohort and sample protocols for this study are the same as those previously described [
10].
4.2. RNA Extraction and Sequencing
Our RNA extraction and sequencing (RNA-seq) protocols for this study are the same as those previously described [
10].
4.3. Differential Gene Expression Analysis
Our bioinformatics approach to analyzing differential gene expression for this study are the same as those previously described [
10].
4.4. Blood Transcription Module Enrichment Analysis
Gene set enrichment analysis was performed using blood transcription module (BTM) gene sets according to the same protocols as previously described [
10]. To summarize these analyses, BTMs were categorized into different families reflecting immune cell types or immunologic processes (as characterized by Braun et al.): B cells, cell cycle, dendritic cell/antigen presentation, type I interferon (IFN type I), myeloid activity/inflammation, T/NK cells, and others [
20]. The percentage of BTMs in each significantly enriched BTM family was then quantified at each time point.
4.5. Group-Level and Single-Subject Blood Transcription Module Network Construction
For BTM network construction, BTMs that demonstrated a significant effect of time point on eigengene expression were selected as candidate BTM nodes. The significance of time point was assessed using an ANOVA with the main effects of group and time point and the random effect of subject. The p-values for the main effect of time point were FDR-corrected across BTMs. Candidate BTMs with significant membership gene overlap were excluded by the following criterion: if candidate BTMs overlapped with a Jaccard index greater than 0.2, then only the BTM with the larger number of membership genes was retained. Using this final set of BTMs, pairwise Pearson correlations were performed between all BTM eigengenes across subjects and V1D0, V1D7, V2D0, and V2D7 samples, separately for each subject group to generate one group HC co-expression network and one HD co-expression network.
Single-subject co-expression networks were constructed in a similar fashion to the group networks, but with only four samples per network (V1D0, V1D7, V2D0, V2D7). Specifically, for each subject, a co-expression network was constructed using the same set of BTMs utilized in the HC and HD group co-expression networks. For a given subject, pairwise Pearson correlations were performed between all BTM eigengenes across all four time points.
4.6. Group-Level Blood Transcription Module Co-Expression Network Comparison
To compare the HC network to the HD network, Fisher’s Z transformation was applied to each network’s edges, and then the Z-transformed HC network was subtracted from the Z-transformed HD network to obtain a Z-score difference network. p-values for the Z-score difference network, calculated from a Z-score to p-value transformation, were FDR-adjusted across edges. Edges that were not significantly different between HD and HC after FDR correction were set to 0 in the Z-score difference network.
In order to characterize the level of coregulation within each given BTM family (and the difference between HC and HD subjects), intrafamily co-expression was investigated. Intrafamily co-expression represents a subnetwork in which nodes comprise all BTMs from a given family (e.g., B cells, cell cycle). For each BTM family subnetwork, we quantified the number of differentially co-expressed edges from the Z-difference network that were (1) positively co-expressed in both HD and HC subjects, but weaker (less positive) in HD, (2) positively co-expressed in both HD and HC subjects, but stronger in HD, (3) negatively co-expressed in both HD and HC subjects, but weaker (less negative) in HD, (4) negatively co-expressed in both HD and HC subjects, but stronger in HD, (5) positively co-expressed in HD subjects, but negative in HCs, (6) negatively co-expressed in HD subjects, but positive in HCs.
These numbers of dysregulated edges were then divided by the total number of possible edges within the BTM family (n choose 2, where n is the number of nodes in the BTM family), yielding the percentage of edges within each family demonstrating each class of differential co-expression. A similar approach was used to characterize the differential co-expression of BTMs between BTM families (interfamily co-expression). For each BTM family, percentages of dysregulated edges between BTMs within a given a family (first node) and BTMs outside of the family (second node) were quantified. Finally, percentages of dysregulated edges were quantified pairwise between the BTM families.
4.7. Single Subject-Level BTM Co-Expression Network Comparison
Edge weight (Fisher Z) distributions for single-subject co-expression networks were compared between HD and HC groups both globally and for each BTM family. A global statistical comparison of edge weights was achieved by quantifying the median positive edge weight per subject, and then comparing these between the HD and HC groups using a Student’s t-test. Global median negative edge comparisons were performed in the same way.
To characterize the differential co-expression of intrafamily BTMs, the median positive edge weight across all edges within a BTM family was calculated on a per-subject basis. These median edge weights were then compared between the HD and HC groups using a Student’s t-test. The median intrafamily negative edge weight within each BTM was compared in the same fashion.
A similar approach was used to characterize the differential co-expression of interfamily BTMs. For each BTM family, the median positive edge weight across all edges between BTMs within a given family (first node) and BTMs outside of the family (second node) were quantified and then compared between the HD and HC groups. The median interfamily negative co-expression was compared in the same fashion.
4.8. Gene Regulatory Network Construction and Analysis
To more specifically characterize the regulatory interactions underlying altered co-expression networks, gene regulatory networks were constructed separately for HD and HC groups using PANDA (Passing Messages between Networks for Data Assimilation) [
21]. For the inputs to the PANDA algorithm, we used (1) an initial TF–gene regulatory matrix derived from the network on the Glass et al. website (
https://sites.google.com/a/channing.harvard.edu/kimberlyglass/tools/resources (accessed on 23 March 2022), which was based on the TFs present in (2) our previously variance-stabilized gene expression matrix, and (3) a protein–protein interaction matrix derived from the STRING database interaction scores between all TFs used in the initial TF–gene regulatory matrix. The output regulatory network for controls was then subtracted from the output HD regulatory network, yielding a regulatory difference network. Gene set enrichment analysis was then performed using the
clusterProfiler package [
64] with BTM gene sets and a list of gene targets ranked by most significant edge differences from the regulatory difference network. BTMs with FDR-adjusted
p < 0.05 were considered significantly enriched. The core enrichment genes, representing those genes that contribute most to the enrichment signal of the BTM, were obtained for the most enriched BTMs.
4.9. Cell Deconvolution Analysis
To determine whether the regulatory findings are driven more by changes in immune cell populations than activity, we performed cell deconvolution analysis on our previously variance-stabilized gene expression matrix at four time points (V1D0, V1D7, V2D0, V2D7) using
CIBERSORTx [
65]. For the deconvolution reference, we constructed a custom signature matrix based on a published single-cell RNA-seq dataset of vaccinated control PBMCs to yield relative proportions for 16 immune cell populations (“CD4 T”, “CD14+ monocytes”, “Naive CD8 T”, “CD8 T”, “CD16+ monocytes”, “NK”, “cDC2”, “B”, “pDC”, “HPCs”, “Platelets”, “NK T”, “Plasmablasts”, “Tregs”, “Naive B”, “cDC1”), which should be more representative of the cell markers and populations expected in our vaccinated cohorts [
36]. Wilcoxon rank sum tests were performed to assess significant differences in relative proportions between the HD and HC groups for each cell type at each time point, as well as between time points for each cell type.