1. Introduction
Neutrophil extracellular trap (NET) formation is a self-defense response in which neutrophils eject DNA strings into the extracellular space [
1]. Upon stimulation with phorbol 12-myristate 13-acetate (PMA) or various pathogen-related materials, such as formyl-methionyl-leucyl-phenylalanine, decondensation of chromatin, and rupture of the nuclear membrane, DNA strings together with various cell proteins, such as histones, neutrophil elastase, and myeloperoxidase (MPO), spread out from the cells [
2]. When DNA strings and these proteins spread in infected tissues, they serve as effective bactericidal nets. Therefore, NET formation acts as a natural self-defense system. However, DNA and histones are well-known damage-associated molecular patterns (DAMPs), and DNA and proteins released from cells also act as inducers of inflammatory responses involved in various diseases [
3], including rheumatoid arthritis [
4], thrombosis [
5], and atherosclerosis [
6].
NETs were initially found as anti-bacterial self-defense system of neutrophils. Currently, NETs are classified into three types depending on the morphological changes in cells: vital NETs, mitochondrial NETs, and suicidal NETs [
7]. In vital NET formations, through stimulation with
Staphylococcus aureus, NETs are released from the cells through nuclear budding and vesicular release. Cells release DNA without disruption of the plasma membrane and cellular death; however, the cells become anuclear [
8,
9]. Mitochondrial NETs are induced through pretreatment of neutrophils with GM-CSF and subsequent stimulation with LPS or complement factor 5a. Under these conditions, neutrophils release mitochondrial DNA, but not nuclear DNA, as extracellular traps [
10]. When neutrophils are stimulated with phorbol 12-myristate 13-acetate (PMA), decondensed DNA is released from the cells through membrane perforation. Such a lytic process of NET formation is called suicidal NETs [
9].
DNA release similar to NET formation was also observed in some granulocytes, such as monocytes and eosinophils [
11]. Thus, the words ETs and ETosis can be used to refer to DNA release from various cells and the cell death associated with DNA release of these cells.
Cholesterol is oxidized either enzymatically or non-enzymatically under physiological conditions to generate various oxysterols [
12,
13]. Bile acids are the major metabolites of cholesterol that are generated in the liver and secreted into the bile juice. In the first step of bile acid formation, cholesterol is hydroxylated to form 7α-hydroxycholesterol (7αHC) [
14]. In the neural tissues, cholesterol is converted to 24-hydroxycholesterol (24HC), which is released into the bloodstream and transferred to the liver [
15]. Cholesterol is also oxidatively modified non-enzymatically through interactions with free radicals, both in vivo and in vitro. It is well-known that the major oxidized product of cholesterol in oxLDL is 7-ketocholesterol (7KC), and it is a cytotoxic product [
16]. Apoptosis of endothelial and smooth muscle cells, but not of human fibroblasts, was induced by 7KC [
17]. Apoptosis of neuronal cells was induced by 7KC through activation of the nuclear factor–kappa B (NF-kB) and Akt pathways [
18]. Some oxysterols, including 24HC and 27-hydoxycholesterol (27HC), are ligands of the liver X receptor (LXR), and 25-hydroxycholesterol (25HC) induces LDL receptors via the activation of the sterol regulatory-element binding protein (SREBP)-SREBP cleavage activating protein (SCAP) system [
19]. Oxysterols were shown to accumulate in atherosclerotic plaques [
20]. In addition, the plasma level of 7KC was reported to be associated with risk of cardiovascular events and total mortality based on a prospective follow-up study [
21].
Several oxidative products have been reported to be involved in enhancing DNA release from cells. For example, oxidized phospholipids, such as oxidized phosphatidylethanolamine [
22] and oxidized phosphatidylcholine [
23], have been reported to enhance NET formation. Oxidized low-density lipoprotein (oxLDL), which is known as a source of oxidized phospholipids in vivo, was shown to enhance PMA-induced NET formation in neutrophils [
24]. In addition to the oxidation products of phospholipids, 7KC and other oxysterols are generated in oxidized LDL [
25], and the cytotoxic effects of these oxysterols have been reported in various cells, including vascular smooth muscle cells [
17,
26]. However, the effect of oxysterols that could affect membrane stability in NETosis has not been fully investigated.
Cholesterol and its metabolites are essential components that affect the membrane stiffness of the cell membrane [
27]. We postulated that membrane components may affect NET formation because disruption of the plasma and nuclear membranes is accompanied by DNA release from neutrophils. Thus, we assumed that oxysterols, such as 7KC, might activate NET formation. In the present study, we examined the effects of four oxysterols on the release of DNA and proteins from neutrophils. Unexpectedly, oxysterols, but not cholesterol, suppressed DNA release from neutrophils. Furthermore, treatment of cells with methyl-β-cyclodextrin (MβCD) that extracts sterol compounds from cell membranes to form inclusion complexes enhanced DNA release from PMA-stimulated cells. These observations suggest that oxysterols may act as modulators of neutrophil activation.
2. Materials and Methods
2.1. Materials
The following biological materials were procured commercially: 7α-hydroxycholesterol (7αHC) was from FOCUS Biomolecules (Plymouth Meeting, PA, USA); 7-ketocholesterol (7KC) and 25-hydroxycholesterol (25HC) were from Cayman Chemicals (Ann Arbor, MN, USA); 27-Hydroxycholesterol (27HC) was from Sigma-Aldrich (St. Louis, MO, USA); deutelium-labeled 7-ketocholesterol-22 (7KC-d7) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Micrococcal nucleases were from Takara Bio Inc. (Shiga, Japan); polyclonal rabbit anti-myeloperoxidase (MPO; A0398) antibody was from Dako (Carpinteria, CA, USA); polyclonal rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; G9545) antibody was from Sigma-Aldrich (St. Louis, MO, USA); SYTOX Green was from Thermo Fisher Scientific (Waltham, MA, USA); the Cytotoxicity LDH Assay Kit-WST was from Dojindo (Kumamoto, Japan); and the MTT Cell Count Kit was from Nacalai Tesque (Kyoto, Japan). All other chemicals were of analytical grade and were obtained from Sigma-Aldrich, Fuji Film Wako (Osaka, Japan), or Nacalai Tesque (Kyoto, Japan).
2.2. Cell Culture and Cell Treatment
Peripheral neutrophils were isolated from freshly drawn blood of healthy donors, as previously reported [
28]. HL-60 cells (obtained from the American-Type Culture Collection) were maintained in Roswell Park Memorial Institute-1640 medium (RPMI-1640) supplemented with 5% (
v/
v) fetal bovine serum (FBS), 100 U/mL of penicillin, and 100 μg/mL of streptomycin. All cell cultures were maintained in a humidified incubator at 37 °C under an atmosphere of 5% CO
2. To differentiate the cells into neutrophil-like cells, HL-60 cells were cultured in the presence of 2 µM of all-
trans-retinoic acid (AtRA) in RPMI-1640 supplemented with 5% (
v/
v) FBS for four days following the procedure described previously [
23]. Differentiation of cells was checked based on increases in the gene expression of
ITGAM and
FCER1A (genes for CD11b and Fcε receptor, respectively,
Supplementary Materials). Because we did not examine specific markers for neutrophil differentiation, we describe the HL-60-derived cells in this study as granulocytes. Differentiated cells were recovered, resuspended in serum-free RPMI-1640, and used in the experiments.
2.3. Measurement of Released DNA
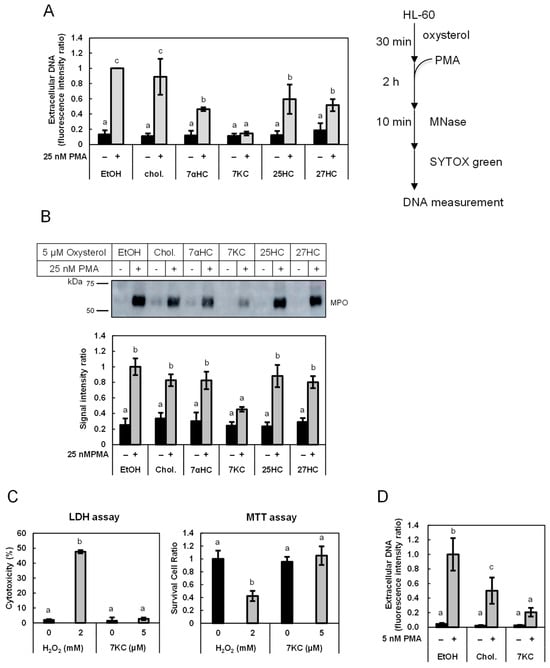
The released DNA was quantified as described in our previous study, with slight modification [
24]. Differentiated HL-60 cells were seeded in a 24-well plate (5.0 × 10
5 cells/well) and incubated for 30 min at 37 °C. Cells were treated with the indicated concentrations of either one of the oxysterols for 30 min, and DNA release was induced through an additional 2 h of incubation with 25 nM of PMA. Human peripheral blood neutrophils were seeded in a 24-well plate (5.0 × 10
5 cells/well) precoated with poly-L-lysine and incubated for 30 min at 37 °C. Cells were treated with 7KC, cholesterol, or vehicle for 30 min, and DNA release was induced through an additional 2 h of incubation with 5 nM of PMA. For the experiments with sterol extraction, up to 100 μM of methyl-β-cyclodextrin (MβCD) was added to the cells 30 min before the beginning of the 2 h of incubation with 25 nM of PMA. Thereafter, the samples were treated with 1 U/mL of Micrococcal nuclease for 10 min at 37 °C to degrade the released DNA. The debris were removed through centrifugation at 1800×
g for 10 min, and the supernatant was mixed with 1 µM of SYTOX Green
®. The fluorescent intensity was measured using Varioskan
® Flash (Thermo Fisher Scientific).
2.4. LC-MS/MS
Differentiated cells were harvested by removing the culture medium with centrifugation at 1500×
g for 5 min. After the cell pellet was washed with phosphate-buffered saline (PBS), the cells were resuspended in PBS and split into two tubes: one for measuring sterol content through liquid chromatography with tandem mass spectrometry (LC-MS/MS) and the other for measuring cellular proteins. After removing the PBS, the cellular lipids were extracted using hexane/2-propanol (3:2,
v/
v). As an internal standard, 600 ng of 7KC-d
7 was added to the samples. Samples were dried under a nitrogen gas stream, and the lipids were re-dissolved in 95% (
v/
v) acetonitrile and centrifuged at 20,000×
g for 15 min to remove insoluble matter. Oxysterols and cholesterol were quantified through LC-MS/MS analysis using atmospheric pressure chemical ionization (APCI), as cholesterol is not efficiently ionized through electrospray ionization (ESI) [
29]. LC-MS/MS was performed using QTRAP6500+ with APCI (SCIEX, Tokyo, Japan) equipped with an InertSustain C18 HP column (3 µm, 2.1 × 100 mm: GL Science, Tokyo, Japan). Five microliters of the sample was injected using an autosampler. Solvent A was ultrapure water containing 0.1% (
v/
v) formic acid, and solvent B was acetonitrile containing 0.1% (
v/
v) formic acid. The column oven was set at 40 °C. The flow rate was 0.2 mL/min. A multistep gradient elution was performed using the following 25 min gradient profile (min/% B): 0 min/75% B–15 min/100% B–23.9 min/100% B–24 min/75% B–25 min/75% B. The APCI source parameters were as follows: source temperature 300 °C, nebulizer current 2, and curtain gas 45 psi. The optimized conditions of mass spectrometry for the detection of cholesterol and oxysterols are listed in
Table 1. Although hydroxycholesterols have the same molecular weight, they can be specifically detected based on the difference in retention time and the combination of precursor and product ions.
For protein content measurement, the PBS-free cells were lysed in lysis buffer (50 mM of Tris–HCl, pH 8.0, 1 mM of EDTA, 150 mM of NaCl, 1% NP-40, and 0.25% sodium deoxycholate). The lysate was centrifuged at 18,000× g for 5 min at 4 °C to remove cellular debris. The amount of protein in the lysate was measured using the BCA assay.
The sterol content was calculated based on the internal standard and normalized to the cell protein.
2.5. Cytotoxicity Determination Through LDH Assay
Differentiated HL-60 cells were seeded into a 24-well plate (5.0 × 105 cells/well) and incubated at 37 °C for 30 min. After the cells were treated with 5 µM of 7KC or 2 mM of H2O2 for 2.5 h, the cell suspension was centrifuged at 250× g for 2 min, and the supernatant was used for determination of lactate dehydrogenase (LDH) released from cells using a Cytotoxicity LDH Assay Kit-WST (Dojindo) according to the manufacturer’s protocol.
2.6. Cell Viability Measurement Through MTT Assay
Differentiated HL-60 cells were seeded in a 96-well plate (2.5 × 104 cells/well) and incubated for 30 min at 37 °C. After the cells were treated with 5 µM of 7KC or 2 mM of H2O2 for 2.5 h, viable cells were determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazoliumbromide (MTT) assay (MTT Cell Count Kit; Nacalai Tesque) according to manufacturer’s protocol.
2.7. Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Statistical significance was calculated using a one-way analysis of variance with Tukey’s post hoc test. Statistical significance was set at
p < 0.05. Easy R (EZR) version 1.61 (The R Foundation for Statistical Computing) was used for statistical analyses [
30].
4. Discussion
In the present study, we found that low concentrations of 7KC and other oxysterols, but not cholesterol, suppressed the release of DNA and MPO in PMA-stimulated HL-60-derived granulocytes. Furthermore, DNA release was accelerated by the removal of (oxy)sterols from the cell membranes following treatment with MβCD. DNA release from human neutrophils was also suppressed by 7KC. These observations suggest that oxysterols suppress PMA-induced DNA release from granulocytes.
Numerous studies have elucidated the significance of NETs, or ETs, in various pathological conditions in vivo, including rheumatoid arthritis, thrombosis, atherosclerosis, and other diseases [
3,
4,
5,
6,
31,
32]. Release of DNA and proteins from granulocytes is currently considered a common phenomenon that induces inflammation; therefore, various compounds that affect granulocytes are potentially useful for understanding and regulating inflammatory responses.
We have previously shown that a small amount of oxLDL enhances NET formation in differentiated HL-60 cells and human peripheral neutrophils after stimulation with PMA [
23]. These observations suggest that oxidized lipids, which are formed in oxLDL as well as in various diseased tissues, may influence NET formation. Oxidized phospholipids containing truncated 2-acyl chains at the
sn-2 position of phosphatidylcholine (truncated oxPC) have been studied as the inflammatory oxidation products of oxLDL [
33]. We showed that 10 μM of truncated oxPC enhances the release of DNA and MPO from differentiated HL60 cells [
24]. Oxysterols, including 7KC, are generated in vivo and thought to be cytotoxic products [
13,
25,
26]. Therefore, it is assumed that the oxysterols may enhance responses like NET formation; however, our observation that oxysterols suppress the release of DNA and MPO in PMA-stimulated neutrophils was unexpected.
Although 7KC was the most effective oxysterol among those tested, it did not show any cytotoxic effects under the experimental conditions. The concentration of oxysterol added to the neutrophils was 5 μM. We assume that this concentration is critical because a previous study [
17] reported that 7KC at 40 μg/mL induces apoptosis and necrosis of vascular cells. This concentration of 7KC (40 μg/mL of 7KC is approximately 100 μM) was 20 times higher than that under our experimental conditions. Tabas et al. reported that 50 μg/mL (125 μM) of 7KC induced NET formation in neutrophils from murine blood [
34]; however, the concentration of 7KC used here was even higher than that used in the previous study. Therefore, 7KC may have a biphasic property depending on the concentration range; 7KC induced DNA release at high concentrations but suppressed it at low concentrations. Furthermore, the effects of a low concentration of oxysterols on the membrane stiffness of endothelial cells were reported previously. Shentu et al. showed that the elastic modulus of bovine aortic endothelial cells increased when exposed to 10 μg/mL of 7KC or 27HC, indicating an increase in cell stiffness [
35]. The effects on membrane stiffness induced by exposure to 7KC or 27HC were reversed through enrichment of the cells with cholesterol [
35]. Oxidized LDL increases the stiffness of cell membranes by supplying oxysterols to and removing cholesterol from cell membranes [
36]. Our results demonstrated that the addition of 7KC suppressed DNA release from granulocytes. Taken together, 7KC under certain concentration might stabilize the cell membranes, resulting in a decrease in NET formation. In this study, we did not examine the effect of oxysterols on vital NETs under a specific condition; however, it is speculated that stabilization of cell membranes by oxysterols could also suppress the exocytotic release of DNA.
In addition, removal of sterols enhanced DNA release in PMA-stimulated cells. MβCD retrieves sterols from membranes and forms inclusion complexes with retrieved sterols [
37,
38]. Oh et al. reported previously that treatment of neutrophils with MβCD reduced the deformability of the membranes [
39]. Such an effect of sterols may explain the alteration of the susceptibility of cells to PMA-induced membrane damage in our experimental conditions. The ligand of MβCD is not necessarily specific for oxysterols; however, because the basal concentration of oxysterols in differentiated HL-60 cells is almost 1000-fold lower than that of cholesterol, the effect of removing oxysterols may be more profound than that of removing cholesterol. These observations suggest that oxysterols act as endogenous suppressors of NET formation.
Previous studies, including ours, showed that some lipophilic compounds, such as resveratrol, suppress DNA release from differentiated HL-60 cells [
40]. Resveratrol was considered to suppress DNA release not through protein kinase C inhibition but rather membrane stabilization because DNA release from cells stimulated with calcium ionophore was also suppressed by resveratrol. Meanwhile, some lipid compounds, such as oxidized phospholipids, increased NET formation [
22,
24]. There might be some characteristics of the molecular structure that are effective in stabilizing membranes, or these suppressive lipid compounds may interact with certain proteins that relate to the NET formation processes.
A comprehensive review summarizes many studies on oxysterols reporting deleterious effects or beneficial effects on inflammation, oxidative stress, cell death, and so on [
41]. For example, 50 μM of 25HC induced IL-8 secretion in U937 human promyelocytic leukemia of neuronal cells; however, 0.1 μM of 25HC decreased LPS-induced IL-1β mRNA and protein and inflammasome activation in cholesterol-25-hydroxylase-defficient bone-marrow-derived monocytes. Generally, beneficial effects are observed with lower concentrations of oxysterols rather than deleterious effects. The responses are diverse, and some of them seem to be indirect responses through signal transduction. Our present observation indicates another unique effect of oxysterols on cellular responses. One possible consideration would be that oxysterols act as physiological regulators at low concentrations under normal conditions but show toxic properties when their concentrations increase to a certain hazardous range. Certainly, more studies are needed to elucidate the behavior and functions of oxysterols in vivo.
A limitation of the present study is that the mechanisms of oxysterol suppression were not elucidated. Oxysterols, including 7KC, are thought to destabilize membranes [
42]. However, we do not think that this is unlikely, because the concentrations used to treat neutrophils were much lower than those used in previous experimental conditions [
34]. Another point is that not all of the cellular responses of ETosis were examined in this study. However, we detected citrullinated histones and shape changes under electron microscopy of differentiated HL-60 cells after PMA treatment in our previous study [
24]. Another point would be that the present study only included a cell culture study. For the next step, an in vitro or clinical study would clarify the effects of oxysterols on physiological and pathological responses. To conclude this issue, the effects of oxysterols on the physicochemical properties of the cellular membranes should be examined. Our observations suggest that oxysterol-sensitive machinery suppresses NET formation by neutrophils. Further work is needed to identify these target molecule(s), which is the next goal of our subsequent study.