1. Introduction
Recent years have seen the development of numerous algorithms and computational packages for the analysis of multiomics data sets [
1,
2,
3,
4,
5,
6,
7]. As with other applications of machine learning, problems addressed by these algorithms are divided into two broad categories: unsupervised (e.g., clustering or class discovery) and supervised (including class comparison and class prediction) [
8]. Advances in the area of unsupervised learning have been substantially broader and deeper than the advances in supervised learning.
One challenge when trying to integrate multiomics data sets arises from multiple levels of missing data. At a low level, individual features in some data sets may fail to be measured in individual samples. It is usually easy to deal with low-level missing data. In some cases, they can be ignored; in others, appropriate imputation methods may be able to fill in the blanks [
9,
10]. At a high level, some patients may have been omitted entirely from one of the omics data sets. A common, highly conservative, response is to work with the subset of patients who have “complete data”, that is, the intersection of samples used in all data sets [
11]. This approach may throw away much potentially useful data. Especially for time-to-event (“survival”) analyses, where the statistical power depends on the number of events, restricting to the complete data subset may reduce the total sample size to a point where it is no longer possible for a machine learning algorithm to learn a useful prognostic model.
As of this writing, we do not know of any supervised multiomics method that can learn to predict outcomes when some samples have only been assayed on a subset of the omics data sets. In this paper, we present a novel algorithm, Partial LeAst Squares for Multiomics Analysis (PLASMA), to predict time-to-event outcomes in the presence of incomplete data. The core idea of PLASMA relies on a two-step process, built on ideas that have proved useful for analyzing related problems in single omics data sets.
Use partial least squares (PLS) with Cox regression analysis [
12] to identify factors that can predict time-to-event outcomes in each individual omics data set.
For each pair of omics data sets, use samples common to both data sets to train and extend models of each factor to the union of assayed samples. For this step, we use PLS with linear regression [
13,
14].
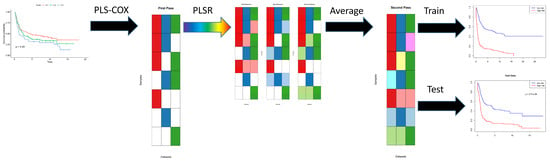
First, for each of the omics data sets, we apply the PLS Cox regression algorithm to the time-to-event outcome data to learn separate predictive models (indicated in red, green, and blue, respectively, in
Figure 1). Each of the individual models may be incomplete, since they are not defined for patients who have not been assayed (shown in white) using that particular omics technology. In the second step, for each pair of omics data sets, we apply PLS linear regression to learn how to predict the Cox regression components from one data set using features from the other data set. This step extends (shown in pastel red, green, and blue, respectively) each of the original models, in different ways, from the intersection of samples assayed on both data sets to their union. Then we average all the different extended models (ignoring missing data) to get a single coherent model of components across all omics data sets. Assuming that this process has been applied to learn the model from a training data set, we can evaluate the final Cox regression model on both the training set and a new test set of patient samples.
Our approach is motivated, in part, by the unsupervised method of Multi-Omics Factor Analysis (MOFA) [
15,
16]. MOFA, like PLASMA, does not require all omics assays to have been performed on all samples. It is still able to discover class structure across omics data sets. Also like PLASMA, it starts with a standard method—Latent Factor Analysis—that is known to work well on a single omics data set. It then fits a coherent model that identifies latent factors that are common to, and able to explain the data well in, all the omics data sets under study. Our investigation (unpublished) of the factors found by MOFA suggests that, at least in some cases, it is approximately equivalent to a two-step process analogous to the one we propose in PLASMA.
Terminology
Because of the layered nature of the PLASMA algorithm, we intend to use the following terminology to help clarify the later discussions.
The input data contains a list of omics data sets.
Each omics data set contains measurements of multiple features.
The first step in the algorithm uses PLS Cox regression to find a set of components. Each component is a linear combination of features. The components are used as predictors in a Cox proportional hazards model, which predicts the log hazard ratio as a linear combination of components.
The second step in the algorithm creates a secondary layer of components. We do not give these components a separate name. They are not an item of particular focus; we view them as a way to extend the first level components to more samples by “re-interpreting” them in other omics data sets.
4. Discussion
One of the most interesting aspects of this study is that the PLASMA algorithm successfully discovered a model on the training set that generalized to produce statistically significant results on the test set (
Figure 4). Further, it accomplished this goal even though most of the individual omics models did not generalize well to the data set (
Supplemental Figure S2). We can think of three possible reasons to explain this performance.
“Wisdom of the crowd”. There has long been an idea in the machine learning field that combining ensembles of weak models can give rise to a strong model. Well-established examples of this idea are bagging and boosting [
24]
“Out of phase”. Each omics data set may overfit the model in a different way. Instead of reinforcing each other, the extent of overfitting may cancel out.
“Feature Elimination”. The combined method successfully identifies useful predictive factors. So, we are still able to fit a generalizable Cox model on the final components.
More research will need to be conducted on a variety of data sets to determine whether this phenomenon is more general.
In the original analysis of the STAD dataset by the TCGA Research Network [
18], the authors identified four molecularly distinct subtypes: tumors positive for Epstein–Barr virus; microsatellite unstable tumors; genomically stable tumors; and tumors with chromosomal instability. These subtypes, albeit possessing distinct molecular characteristics, were not significantly different from each other in terms of survival outcomes. We also performed an unsupervised analysis using MOFA, and found that the latent factors it uncovered were not related to survival (
Supplemental Tables S1–S3, Supplemental Figures S8–S13). Thus, we took a different approach: we trained a multiomic model supervised on overall survival, and then dissected the feature weights of omics assays.
First, however, we confirmed that the PLASMA model for STAD could be validated in an independent cohort. We divided the TCGA-ESCA data into esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC). Based on known biological similarities between EAC and STAD [
17,
23], we hypothesized that external validation of the PLASMA STAD-model with EAC would yield positive results, and validation with ESCC would serve as a negative comparison. Indeed, we observed that the Kaplan−Meier plot of OS shows little separation between the low- and high-risk patient groups in the validation with ESCC, while there is significant separation in the validation with EAC (
Figure 5).
We recognize that a prognostic or predictive model that depends on multiple genome-wide omics technologies is unlikely to move into clinical use soon. The costs to perform the assays, both in dollars and in the delays imposed by waiting for all results, limit its clinical applicability. We expect that it will be necessary to refine the model to use a much smaller set of molecular predictors before it can be clinically useful. Toward that eventual goal, we performed several analyses to determine where to focus our attention.
One approach involved performing gene enrichment analysis to understand the biological role of the complicated factors that PLASMA found across omics data sets (
Figure 7). Somewhat surprisingly, the final gene set had weaker associations with any of the biological pathways than did the individual components derived from single omics datasets. However, a heatmap of the same values (
Supplemental Figure S3) indicates that the annotation categories for the final gene list are strongly correlated with those for the union of all the component gene sets. It is possible that the associations are weaker because the final model retains fewer related/redundant genes than the individual PLS models. Future research in other disease contexts will be required to understand this phenomenon.
Nevertheless, the gene enrichment analyses may provide a useful way to focus on important factors for overall survival in gastric cancer. The left and right ends of the bubble plot (and the heatmap) mainly contain pathways that distinguish between the factors. But the central portion of the plots, from the KEGG_ERBB_SIGNALING_PATHWAY on the left to the WP_GLIOBLASTOMA_SIGNALING_PATHWAYS on the right, primarily consists of signaling pathways that have been identified as important across all the multiomic factors we found. Moreover, many of these signaling pathways have already been identified in the literature as important in gastric cancer. These include signaling pathways involving ERBB (EGFR) [
25], Gastrin [
26], Leptin [
27], KIT [
28], Oncostatin M [
29], CKAP4 [
30], and the PI3K-AKT-MTOR pathway [
31]. There is also a strong association with Prolactin signaling, which has previously been highlighted for its role in breast cancer [
32] and in colon cancer [
33].
One limitation of gene enrichment analysis is that it ignores the direction of association between pathways (or genes) and survival. Genes with both positive and negative weights, either for individual components or in the final model, were combined when performing gene enrichment analysis. In order to better understand the contributions of individual gene features, we looked more closely at their weights in the final model of survival (
Figure 6). We found that some of the binary clinical variables (ClinicalBin) successfully separated high- and low-risk patients in the STAD test set. Furthermore, the weights given to these features in the final composite model appeared reasonable. Being tumor free had a large negative weight, indicating decreased risk, while having a residual tumor had a positive weight, indicating increased risk. Similarly, having grade 2 or stage IB tumors or complete response to therapy were markers of good prognosis, while having grade 3 or stage IIIB tumors were markers of poor prognosis.
From the mutation allele format (MAF) dataset, we found that the most positive weight is from
ERBB3, a member of the
EGFR family that can heterodimerize with other family members, notably
ERBB2 [
34]. Overexpression of
ERBB3 and
ERBB1 mRNAs and proteins in histological specimens of gastric cancer has been significantly associated with tumor relapse and poorly differentiated morphology [
35]. From the RPPA data set, we observed that ERBB2 possesses a positive weight, and is therefore positively associated with worse outcome. This result, again, matches what is currently known in
ERBB2-mutant gastric cancers and other cancers such as breast and lung cancers: overexpression of ERBB2 promotes tumor growth and survival by downstream signaling through the PI3K-AKT and RAS-MAPK pathways. Many of the results from the mRNAseq data set can also be validated from the literature. These include the fact that increased expression of
SYK [
36],
DMBT1 [
37],
CASP7 [
38],
OLFM4 [
39], and
PIGR [
40] has been reported to be associated with prolonged survival. High expression of
MAGEA6 [
41],
DKK1 [
42], and the MYC-induced long noncoding RNA
H19 [
43] has been associated with shorter survival.
In a wide variety of cancers,
TP53 mutations are generally associated with a poor prognosis, including in gastric adenocarcinomas when
TP53 was assessed by immunohistochemistry [
44]. However, our analysis of the MAF data set found that the presence of a
TP53 mutation was associated with a negative weight, and therefore better overall survival. This result is supported by a univariate analysis using the STAD TCGA data set that reported that wild type
TP53 was associated with poorer prognosis in patients with gastric cancer [
45], confirming independent findings reported previously [
46,
47,
48]. However, several other studies found no association between
TP53 and survival in stomach cancer [
49,
50,
51,
52]. Earlier studies were characterized by a variety of methods to assess mutation status, including immunohistochemical staining [
44] and limited direct sequence analysis of specific mutation hotspots [
53]. Since our approach is based on whole exome sequencing of the entire gene, as well as the indirect contribution of genes such as
TP53 through various components, it is still possible that
TP53 has a small, non-independent effect that merely contributes to the effects of other mutations. We also found that
ATM mutations and
ATM expression were associated with a better outcome. Similar to mutated
TP53, the literature contains conflicting results, with reports of low
ATM expression associated with both poor [
54] and good [
55] prognosis. These apparently discrepant findings suggest that the association between mutation status and prognosis depends upon both the context and the methods used to assess mutation status.
The weights from the miRNA-seq data set have mixed agreement with what is known about the role of miRs in STAD. We found that miR-509 and miR-338 have negative weights and thus lower levels should be associated with worse prognosis, which agrees with the literature [
56,
57,
58,
59]. But we also found that miR-552, miR-105, and miR-196a have negative weights, yet the literature suggests that higher levels of these miRs are associated with tumor severity in gastric cancer or breast cancer [
60,
61,
62]. Similarly, downregulation of miR-31 has been reported to be associated with worse overall survival of gastric cancer patients [
63], which, again, is the opposite of our results. We are unable to fully explain the discrepancies between our findings and the literature. The differences may be related to the fact that many earlier studies used different technologies (for example, microarrays rather than sequencing). There may also be differences depending on whether assays are targeting primary miRNAs, precursor miRNAs, or 3′ or 5′ mature miRNAs.