1. Introduction
Functional three-dimensional protein structures are maintained dynamically via intramolecular interactions and protein‒protein interactions [
1,
2]. Such structure‒function relationships in proteins are important for understanding the molecular mechanisms of protein activities and rationally developing protein inhibitors for biochemical research and medicine. Protein functional sites may include substrate recognition sites, catalytic active sites, allosteric sites for conformational change, protein‒protein interaction sites, and other important sites to achieve specific structures and functions. Among the various approaches to understanding protein molecules and discovering novel drugs, peptides and their derivatives have been proposed to be useful, and this approach is known as peptidomimetics [
3,
4,
5,
6,
7,
8,
9]. For example, combinatorial synthetic peptide arrays and libraries may be used to capture low-abundance proteins, understand protein‒ligand interactions, and develop novel peptidomimetic drugs [
10,
11]. These peptide-based techniques may be useful, especially for peptide receptors, including some G-protein-coupled receptors [
12], but a similar idea can be applied not only to peptide receptors but also to any protein in general.
Among various intramolecular and intermolecular interactions, protein‒protein interactions (PPIs) are considered excellent targets of peptide ligands for drug development [
12,
13,
14,
15,
16,
17]. For example, α-helix-mediated PPIs may be targeted for potential inhibitory sites in various proteins [
18]. In contrast, a β-sheet region of the HIV protease has been targeted to inhibit the dimerization of this protein by peptide ligands [
19]. PPI inhibitors may be screened through phage-displayed peptide libraries [
20,
21]. These peptide ligands may also be designed rationally in silico in reference to three-dimensional structures of potential binding sites [
22,
23,
24,
25,
26,
27,
28]. For example, recent developments in CIP (cancer immune checkpoint) peptide inhibitors have been performed computationally and screened through random peptide libraries [
7]. PPI inhibitory peptides may be designed from the interacting short constituent sequences (SCSs) of target proteins, but the isolated “endogenous” peptides do not usually fold into secondary structures. To make endogenous peptide inhibitors more efficient, peptide conformation should be constrained by incorporating functional groups or artificial backbone into peptides [
29].
There are many three-dimensional structural data of proteins available in the Protein Data Bank (PDB) [
30,
31], and in many cases, major functional sites of proteins have been structurally visualized. These data provide the foundations for understanding protein functions and rational drug design. Furthermore, even if three-dimensional structural data are not available in the PDB, the primary amino acid sequences in proteins may be used to predict three-dimensional structures and functional sites with high precision in silico via an AI-based machine called AlphaFold [
32], then AlphaFold 2 [
33] and AlphaFold 3 [
34], on the basis of which potential inhibitors may be designed rationally. However, experimental validation of functional sites in vitro is still important. To do so, site-directed mutagenesis [
35,
36] may be the method for validating functional sites at the amino acid level. Saturation mutagenesis at several candidate sites may often be needed. However, such mutagenesis experiments are demanding because there are almost infinite possibilities if all sequences are to be covered combinatorially, even though structural predictions of proteins certainly help researchers narrow down candidate sites. A more fundamental disadvantage of conventional methods is that even if a functional site is discovered and examined in detail via these conventional methods, there may be other functional sites yet to be discovered in a known protein. Moreover, protein functional sites may be intrinsically disordered [
37,
38,
39,
40,
41]. In that case, the conventional methods above certainly miss intrinsically disordered functional sites because no rigorous three-dimensional structures are assigned to intrinsically disordered regions.
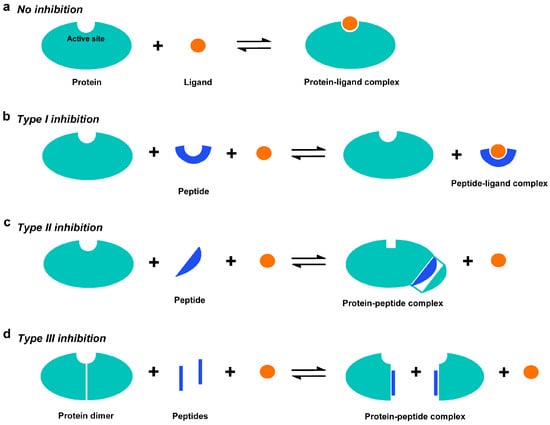
This study presents a peptide inhibitor assay (PIA) for allocating functionally important accessible sites throughout a protein molecule. PIA is a simple, fast, and exhaustive method for investigating functional inhibition by synthetic endogenous peptides (called blocking peptides or BPs in this study) that are derived from short constituent sequences (SCSs) of a target protein itself. The idea of PIA has been derived theoretically as described below. A protein generally forms a protein‒ligand complex to execute its function (
Figure 1a). When a peptide has an amino acid sequence identical to a functional site of a target protein, this peptide may partially mimic the functional site and competitively bind to ligand molecules, consequently lowering the enzymatic efficiency (
Figure 1b). This type of peptide inhibition may be called type I. Because a functional site is often composed of several amino acid residues from different linear positions, type I inhibition by a single peptide is expected to be rare. When a peptide has an amino acid sequence important for three-dimensional structure and functional integrity, intramolecular interactions between amino acids may be competitively inhibited by the binding of the peptide to the corresponding amino acid sequence of the target protein, consequently lowering the functional integrity of the protein (
Figure 1c). This type of peptide inhibition may be called type II. In that case, the competitive sites may not necessarily be catalytically active sites, but nonetheless are important for protein folding and functions. Type II inhibition may be found in peptides corresponding to β-sheet structures due to hydrogen bonds connecting two adjacent strands, but peptides corresponding to α-helices interacting with other parts of the same protein may also function as type II inhibitors. When a peptide has an amino acid sequence identical to the dimerization interface, this peptide may competitively bind to that site, consequently inhibiting dimerization and catalytic functions if dimerization is critical for catalytic activity (
Figure 1d). This type of peptide inhibition may be called type III, and it is this type III inhibition that is an active research field for developing PPI inhibitors, as mentioned above. In any case, if a synthetic peptide shows an inhibitory effect, then the amino acid sequence used for that peptide is considered a candidate functional site or structurally important site in the protein. The type II and type III peptide inhibitors do not compete for active sites in most cases, but they may also be considered “competitive” inhibitors in a broad sense.
Because peptides do not stably fold to mimic endogenous secondary structures, the inhibitory effects of endogenous peptides are likely limited and difficult to detect under the optimum conditions for proteins of interest. Thus, to detect an inhibitory effect, PIA may be performed under conditions that are not optimal for the proteins of interest. Nonetheless, when a high concentration of an endogenous peptide is used, a portion of the endogenous peptide molecules may probabilistically be able to form endogenous secondary structures for a very short time even if that structure may be thermodynamically unfavorable in the steady state. As long as these considerations are met, PIA may be practically attainable. The validity and significance of the inhibitory effects under nonoptimum conditions may be arguable, but the PIA results under nonoptimum conditions should be considered a first step in identifying functional sites, and thus in the development of novel drugs. Another support for PIA comes from a group of studies that stresses the important contribution of short constituent sequences (SCSs) of proteins to secondary structures and the functionality of proteins [
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54]. A well-known structural prediction approach, ROSETTA, is also based on data collection of SCSs from the PDB [
55,
56].
Importantly, PIA does not require any three-dimensional information to investigate candidate functional sites. Equally importantly, PIA can comprehensively cover the overall amino acid sequences of a protein throughout a protein chain, which may be difficult, if not impossible, in mutagenesis-based methods. PIA may be improved to be a high-throughput system once an enzymatic assay system is established. This method can identify only sites accessible for peptides, and nonaccessible sites may not be discovered via this method. For the purpose of screening candidate drugs and epitopes for neutralizing antibodies, this accessibility bias may be convenient for researchers because only accessible sites are valid for functional inhibition by drugs. In any case, PIA may be a starting method for quickly searching for candidate functional sites in proteins.
In this study, we focused on the well-known restriction endonuclease EcoRI as a model protein system [
57]. EcoRI is a type II restriction endonuclease that recognizes the short palindromic DNA sequence GAATTC and cleaves the recognition site in the presence of Mg
2+ ions as homodimers [
58]. It is one of the most common restriction enzymes used for the molecular cloning of DNA fragments. EcoRI, like other type II restriction endonucleases, binds to DNA nonspecifically and scans DNA for specific binding sequences through linear diffusion. This linear diffusion with nonspecific binding to DNA is faster than diffusion proper, and is thus called facilitated diffusion [
59]. Facilitated diffusion seems to have two parts, sliding and hopping, and the latter requires high ionic strength [
60], which may be assisted by a quantum walk mechanism [
61]. After facilitated diffusion, specific binding to GAATTC induces a conformational change in EcoRI and DNA, resulting in the activation of the catalytic reaction [
62].
To allocate functional sites, as is the case for other proteins, the crystal structures of EcoRI [
62,
63] have been the basis for site-directed mutagenesis [
64]. Amino acid sequence of EcoRI with secondary structures and known functional sites are shown in
Figure 2. Crystal structures indicate that R200 binds to guanine and that E144 and R145 form hydrogen bonds with the adenine of the cognate DNA sequence [
64]. Indeed, site-directed mutagenesis confirmed that catalytic activity, but not specificity, decreased in E144Q and R145K [
65]. Similarly, the specificity of mutants with R200K, E144Q + R145K, and E144Q + R145K + R200K did not change, but the catalytic activity decreased [
66,
67,
68]. Furthermore, D59 is not important for catalysis [
69]. M137 functions for specific DNA recognition but not I197 [
70]. Site-directed mutagenesis at Q115 [
71] and at P90, D91, E111, and K113 at the catalytic center has been shown to be essential for catalysis [
72]. Temperature-sensitive mutants generated via random mutagenesis have been analyzed: G78D and A235E affected the protein–protein interaction (PPI) for dimerization, and P90S affected the active site [
73]. The promiscuous mutant A138T binds more tightly to the cognate DNA sequence than does wild-type EcoRI but has higher star activity at AAATTC [
74,
75]. H114Y and its saturation mutagenesis revealed that H114 contributes to both recognition and catalysis [
76]. Additionally, N141 has been shown to be essential for specific cognate DNA recognition [
77]. On the other hand, the N-terminus of EcoRI has not been visualized in crystal structures. Solution structures revealed conformational changes in EcoRI when binding to DNA and Mg
2+, and that the N-terminus plays an important role in dimerization [
78,
79].
While the structure‒function data on EcoRI have accumulated over decades, as mentioned above, there seem to be no systematic studies that have scanned the entire EcoRI protein chain for possible functional importance. Importantly, dimeric PPIs and specific binding to the cognate DNA sequence of EcoRI can be inhibited by peptides designed from an α-helix region called α4 [
80,
81]. To characterize EcoRI further, PIA was performed to cover the entire protein chain of EcoRI in the present study. The objective of the present PIA is to examine all possible functional sites with high accessibility, although roughly, throughout an EcoRI molecule via multiple endogenous peptides. The present PIA results were compared with the currently available knowledge of the functional allocation of EcoRI.
Figure 2.
Amino acid sequence of EcoRI (M1-K277). The first M is removed after translation in this protein. The secondary structures and functional sites are indicated below and above the EcoRI sequence, respectively, according to Kim et al. (1990) [
63], Heitman (1992) [
64], and Watrob et al. (2001) [
78]. The blocking peptides (BPs) designed and tested in the present study are indicated below the EcoRI sequence. Major active site sequences are indicated in pink letters (SIKPDGGIVEVKD and WRVVLVAEAKHQG). Red asterisks indicate important sites identified via site-directed mutagenesis. A red dot (E144) indicates salt bridge formation. A sequence of EcoRI corresponding to a helical peptide (α4) that has been demonstrated for its inhibitory effect on EcoRI by Brickner and Chmielewski (1998) [
81] is indicated in red letters (AIERSHKNISEIANFM). Two parallel helices, α4 and α5, that form the inner and outer helices, respectively, to hold DNA are indicated.
Figure 2.
Amino acid sequence of EcoRI (M1-K277). The first M is removed after translation in this protein. The secondary structures and functional sites are indicated below and above the EcoRI sequence, respectively, according to Kim et al. (1990) [
63], Heitman (1992) [
64], and Watrob et al. (2001) [
78]. The blocking peptides (BPs) designed and tested in the present study are indicated below the EcoRI sequence. Major active site sequences are indicated in pink letters (SIKPDGGIVEVKD and WRVVLVAEAKHQG). Red asterisks indicate important sites identified via site-directed mutagenesis. A red dot (E144) indicates salt bridge formation. A sequence of EcoRI corresponding to a helical peptide (α4) that has been demonstrated for its inhibitory effect on EcoRI by Brickner and Chmielewski (1998) [
81] is indicated in red letters (AIERSHKNISEIANFM). Two parallel helices, α4 and α5, that form the inner and outer helices, respectively, to hold DNA are indicated.
4. Discussion
In the present study, we focused on a model protein, EcoRI, to evaluate the feasibility of PIA to characterize functionally important accessible sites in a protein chain. The original 25 blocking peptides (BPs) were designed in reference to secondary structures to cover the entire protein chain without critical information on functional sites. In this way, information on secondary structures is helpful for designing BPs, but PIA can be performed without such information. In fact, PIA does not need any three-dimensional information. Intrinsically disordered regions can also be examined. Moreover, PIA can exhaustively examine the entire sequence of a protein chain. Thus, PIA can be considered a hypothesis-free test. Alternatively, PIA can be used in accordance with a hypothesis on the basis of previous PIA results or other information on functional sites. The PIA results are likely influenced by the accessibility of the peptides to the corresponding sequences and cannot completely identify “functional” sites in a strict sense. This also means that the PIA results reflect accessibility automatically, which may be convenient for identifying drug candidates. The discovery of unknown functional sites in a broad sense may be expected in the course of PIA, as shown in the present study. To execute PIA, the reaction conditions for a protein of interest are important. The present study used nonoptimum reaction buffer, an incubation time of 3 h, and an arbitrary 1:500 concentration of BPs on the basis of the initial assessment. Such a search for reaction conditions is important for PIA to work properly.
Among the 79 endogenous peptides tested in the present study, the original set of peptides worked better than the staggered peptides and short peptides did, demonstrating that secondary structures are indeed functional units of intra- and intermolecular interactions and may be considered recommended units of BP design. Indeed, the nine most efficient peptides largely corresponded to secondary structures (
Figure 9). An exception was BP16, which is located between β4 and βiii (
Figure 2). Site-directed mutagenesis studies have revealed several amino acid residues in the middle region of EcoRI for catalytic function or DNA recognition [
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77], but this region was largely not inhibitory in the present study. The major active site, which is composed of α4 and other secondary structures, was not inhibited very much by BPs, possibly because of low accessibility. These results may challenge the conventional view of rational drug design that mainly targets active sites. Rather, according to the present study, functional sites that differ from active sites may be promising for the development of novel peptide drugs.
Randomized peptides of BP7, BP10, and BP13 showed mixed results: BP7R2 and BP13R showed lower inhibition (higher EcoRI activity) than the original peptides did, which is theoretically expected if the inhibition is sequence specific, but BP10R1 and BP13R1 showed greater inhibition, which is theoretically unexpected. The reason for these results is not clear, but these perplexing results suggest that the inhibitory effect may not be completely sequence-specific and that amino acid contents are also important, at least in these three cases. Correlation analysis revealed the importance of aromatic residues (F, H, W, and Y) for inhibitory effects, supporting the idea that amino acid contents are also important. Surely, other results suggest the importance of sequence specificity. The validity and interpretation of these data should be clarified in the future.
Although the results of the randomized peptides were difficult to interpret, this experiment is an example of a “mutagenesis” experiment for functional sites. In addition to randomized sequences, many types of mutagenesis experiments are readily possible in PIA: point mutations, domain swapping, and indels. Longer or shorter peptides of various sequences corresponding to protein sequences are possible. In designing blocking peptides, the size and position of windows for short constituent sequences (SCSs) to be covered by peptides may be changed at the discretion of researchers. For example, to perform an exhaustive scan, an SCS window in a protein chain can be slid one at a time from the N-terminus to the C-terminus. Staggered peptides are not necessary in that case, making the PIA truly hypothesis-free in terms of the position of functional sites. To analyze protein activity, the present study used the fluorescence intensity of DNA fragments as output data, but in the future, other methods that can be measured in a high-throughput manner, such as colorimetry based on ELISA (enzyme-linked immunosorbent assay), are favored. Peptide arrays for PIA may be challenging because of the low affinity of BPs for the target protein. Combinatorial addition of two or more peptides in a reaction mixture at the same time may hasten the assay to narrow down the candidate inhibitors.
The short peptides designed from the original peptides used in the present study are indeed like deletion mutants and might have been too short to be effective as an inhibitor of EcoRI. These peptides may be too short to mimic the corresponding secondary structures or bind stably to the target sites, although an exception was BP15C, which was more inhibitory than the original BP15 peptide. In accordance with this view, correlation analysis revealed that the number of amino acid residues (peptide length) and molecular weight were negatively correlated with the relative fluorescence intensity. In other words, peptide length and molecular weight were positively correlated with the inhibitory effects of BPs. Peptide length and molecular weight are directly proportional to approximate volume of peptides [
88]. These results are theoretically expected, considering that the inhibition process is sequence-specific and structure-specific but may contradict the unexpected results of the randomized peptides. On the other hand, the number of aromatic residues (F, H, W, and Y) was negatively correlated with the relative fluorescence intensity. In other words, the number of aromatic residues was positively correlated with the inhibitory effects of BPs, suggesting the importance of amino acid contents. In contrast, the isoelectric point, mean hydrophobicity, aliphatic index, the percentage of hydrophilic residues, instability index, and net charge at pH7 were not correlated. It is interesting to investigate the role of aromatic rings in the inhibition process in the future.
Unexpected results were also obtained in BP19.5, which probably induced nonspecific nuclease activity of EcoRI. Alternatively, BP19.5 itself may have nuclease activity, but this is unlikely because the amino acid sequence of EcoRI corresponding to BP19.5 is not known to have nuclease activity. BP19.5 contains a cysteine residue (C218), which may form an intermolecular disulfide bond and could contribute to nonspecific nuclease activity of EcoRI. However, both BP19.5N (not containing C218) and BP19.5C (containing C218) induced such nuclease activity, suggesting no direct contribution of C218 to the nonspecific nuclease induction. Similarly, BP20 also contains C218 but does not induce nuclease activity, although BP20 has a strong inhibitory effect. Rather, N216 and L217 together with their either N-terminal or C-terminal regions may play an important role in the nonspecific nuclease induction because these two residues were contained in both BP19.5N and BP19.5C, although BP19 did not have such an induction ability. In any case, BP19.5 can be considered a specificity/activity modifier of EcoRI instead of an inhibitor. These unexpected results are perplexing but may lead to unexpected discoveries, which could be considered advantages of the hypothesis-free analysis of PIA.
Mechanistically, if BP19.5 does not bind to substrate DNA and degrade it nonspecifically, there was no type I inhibitory peptide in the present study. Most BPs are likely type II inhibitory peptides. Among these type II inhibitory peptides, the possible active site inhibitors, including Mg
2+-binding sites, were BP8, BP8.5, BP9, BP10, BP10.5, BP13, BP12.5, BP13.5, BP17, BP17.5, BP18, and BP18.5, according to Heitman (1992) [
55]. Except for BP18, these BPs were not very inhibitory. The peptides of the possible dimerization inhibitors were BP1, BP1.5, BP2, BP6, BP6.5, BP7, BP7.5, BP8, BP8.5, BP20, BP20.5, and BP21. They may be classified into Type III inhibitory peptides or PPI inhibitors. Notably, the intrinsically disordered N-terminal region was inhibited by BP1 and its adjacent BP2, which is consistent with the view that intrinsically disordered portions can be targeted for novel drugs [
89,
90]. Despite various levels of inhibition, it is surprising that many peptides were more or less inhibitory. The noninhibitory peptides included BP8, BP5.5, BP18.5, BP7N, BP9C, BP10N, BP13M, BP14N, and BP14M.
The most effective BPs were BP25, BP20, BP5, BP18, BP2, BP15C, BP3, BP25, and BP16 (
Figure 9). Among these nine BPs, BP18 covers α5, a part of the active site. This is the only exception, among the nine most effective BPs, that correspond to the active site, although it is still outside the major active sites (βii-β2 and β3). BP20 and BP2 likely inhibit dimerization sites, as shown in the solution structure studies of Watrob et al. (2001) [
78], as type III inhibitors. These are PPI inhibitors, demonstrating the feasibility of using PPIs as drug targets. In contrast, the PPI inhibitor that has been examined corresponding to α4 in a previous study (BP14 in the present study) [
81] showed only a moderate inhibitory effect. The other BPs among the nine most effective peptides may work as type II inhibitors, which have not been predicted in previous studies.
To discuss individual BPs with high inhibition, any of the three consecutive peptides (BP2, BP2.5, and BP3), but not their flanking peptides (BP1.5 and BP3.5), worked as α1 inhibitors. These three peptides share the VIGI sequence, which may be the core target of functional inhibition. In contrast, the highly inhibitory effect of BP5 disappeared in its flanking peptides, BP4.5 and BP5.5, suggesting that the central S54 in BP5 may be the core target of BP5-mediated inhibition. Likewise, the highly inhibitory effect of BP16 was not observed in the flanking BP15.5 and BP16.5 regions, suggesting that the loop region between β4 and βiii (GSNFLTE) instead of secondary structures may be an important region for BP16-mediated inhibition. The highly inhibitory effect of BP18 was not observed in the flanking BP17.5 and BP18.5, suggesting the functional importance of the N-terminal portion of α5 and its connecting loop, which contains previously identified functional sites (I197, R200 and R203). In the case of BP20, which showed high inhibition, the flanking BP19 and BP20.5 did not show high inhibition, and BP19.5 was an inducer of nuclease activity. Thus, not only βv but also its N-terminal and C-terminal amino acid residues may be important for BP20-mediated inhibition. Finally, for high inhibition by BP25, the full length of αii appears to be required because BP24 did not work well. Together, many of these potential “functional” sites are newly identified, and they may be examined further with additional BPs and with other methods, such as site-directed mutagenesis, in the future, potentially leading to the development of novel drugs. Alternatively, the identified sites may be used for raising neutralizing antibodies as antigenic peptide epitopes. In that case, the accessibility of target regions for antibodies may be high because PIA requires high accessibility of target regions for BPs.
An important question is how some of these BPs can be modified chemically to make protein‒peptide interactions more specific and more stable. To do so, various chemical modifications of peptides may be tested on a trial-and-error basis, but realizing stable secondary structures in peptides may be an important direction for a chemical modification strategy. Replacing peptide bonds with stable nonpeptide structures may be performed as peptidomimetics. EcoRI inhibitors may not be useful drugs in terms of experimental and clinical applications. However, similar strategies involving human or pathogenic proteins may have potential clinical applications. For example, the spike protein of SARS-CoV-2, which has been already analyzed based on SCS distributions [
91,
92,
93], may be subjected to PIA for novel drugs against SARS-CoV-2 infection. SCS-based epitopes of spike proteins for effective neutralizing antibodies may also be identified with the help of PIA. Indeed, peptides and peptidomimetics have been considered excellent candidates for novel drugs for COVID-19 treatment [
94,
95,
96]. Proteins from experimental organisms may also be helpful for understanding the molecular functions of target proteins. For clinical use, peptide inhibitors should be chemically stable and well-tested for side effects. In contrast, for use in experimental animals, in vivo bioassays may be performed directly using relatively high concentrations of targeting peptides to knock down a target protein like an antibody. For example, in our previous study, direct protein delivery to butterfly wing epithelial cells appeared to be more efficient for smaller proteins [
97,
98]. Peptides may be administered instead of antibodies in vivo for direct delivery to cells, which may lead to a novel method for studying protein functions in vivo.