1. Introduction
The hair coat is a unique keratinized skin appendage of mammals consisting of numerous independent hair follicle units. Based on morphological characteristics of hair follicles, they can be classified into primary and secondary hair follicles. The hair coat is a typical product of adaptive evolution, providing crucial functions such as insulation, sensation, and mechanical protection for animals to survive in harsh environmental conditions [
1]. A typical hair follicle comprises microstructures such as the hair bulb, hair shaft, inner root sheath, arrector pili muscle, and sebaceous glands, with various cell types participating in the regulation of hair follicle development and homeostasis [
2,
3]. Hair follicles exhibit self-renewal properties, and a complete hair follicle cycle includes the anagen, catagen, and telogen phases [
4,
5]. An increasing number of studies are aiming to identify the driving factors of hair follicle renewal. In recent years, the role of autophagy in regulating hair follicle developmental status has gradually been unveiled. Studies have shown that both direct and indirect inhibition of the mTOR signaling pathway can induce autophagy to accelerate hair regeneration, while direct induction of autophagy through agonists can also promote hair growth. Furthermore, the level of autophagy during the anagen phase of the hair follicle cycle is higher than that in other phases, suggesting that autophagy promotes hair follicle development [
6].
Autophagy is a physiological mechanism by which cells self-degrade and recycle their intracellular components. The classic autophagy model includes two crucial steps: the formation of autophagosomes that encapsulate the substrates to be degraded and the fusion of autophagosomes with lysosomes [
7,
8]. Autophagy can be classified based on its occurrence mode into macroautophagy, microautophagy, and chaperone-mediated autophagy; it can also be categorized based on the selectivity of degraded substrates into selective autophagy and non-selective autophagy [
9,
10,
11]. Autophagy plays a dual role in the regulation of homeostasis: appropriate levels of autophagy help clear aged and useless intracellular substances, maintaining stability, whereas excessive autophagy may lead to over-clearance of cellular contents, thereby inhibiting cellular function [
12,
13,
14,
15]. Studies have shown that autophagy can affect the viability of hair follicle stem cells and promote hair regeneration by regulating glycolysis [
16]. In addition, autophagy mediates cellular remodeling during the terminal differentiation of epidermal and skin keratinocytes [
17]. Autophagy also plays a significant role in controlling stem cell activation induced by apoptosis and skin inflammation [
18]. Evidence from organ cultures of human scalp hair follicles also indicates that autophagy is crucial in maintaining the regulatory processes of hair follicle development [
19]. The impact of autophagy on hair follicle development also seems to exhibit duality, with studies showing both positive promotion of hair follicle development and negative effects on it [
18]. This may be related to the specific developmental stage of the hair follicle and the particular cell types involved. Hair follicle stem cells are a type of adult stem cell. When hair follicles are in the catagen and telogen phases, hair follicle stem cells are mainly distributed in the bulge region where the arrector pili muscle intersects with the outer root sheath, and these stem cells are in a slow-cycling state. When hair follicles enter the anagen phase, hair follicle stem cells initiate proliferation and differentiation, migrating along the hair root sheath to complete the reconstruction of the hair follicle [
20,
21,
22,
23]. The cyclic activation of hair follicle stem cells is the driving force behind the cyclic reconstruction of hair follicles, and their characteristics make them the most important cell type in hair follicle development.
RORA is a ligand-dependent transcription factor that plays a crucial role in a series of physiological and pathological processes, including circadian rhythm regulation, metabolic regulation, inflammation, and immune system modulation [
24,
25,
26,
27]. When RORA was first discovered, its ligand was not clearly identified, and thus, it was classified as an orphan receptor. However, as related research has progressed, there is a perspective that RORA serves as the nuclear receptor for melatonin [
28,
29,
30]. In recent years, there have been many differing views regarding the ligands of RORA. Some studies suggest that RORA may be a natural receptor for sterols and vitamin D derivatives [
31]. RORA consists of a ligand-binding domain, a hinge region, and a DNA-binding domain. When activated by ligands, RORA can regulate the transcription levels of downstream target genes through monomeric or dimeric forms [
32,
33]. Fortunately, the discovery of synthetic agonists (such as SR1078) and inhibitors (such as SR3335) of RORA has greatly accelerated the progress in research on the functions and physiological effects of RORA. Studies have shown that RORA is highly correlated with conditions such as exercise, spatial cognition, and Parkinson’s disease [
34,
35,
36]. Other studies have shown that RORA exhibits critical regulatory effects in cell apoptosis, epithelial–mesenchymal transition, and oxidative stress [
37,
38,
39]. It is particularly noteworthy that RORA is considered to be highly involved in the regulation of circadian rhythms. Studies have shown that RORA affects circadian rhythms by regulating key circadian rhythm factors such as BMAL1 and CLOCK. Mice with RORA knockout exhibit significant disturbances in their metabolic circadian rhythms [
40]. In cancer research, overexpression of the MYCN gene disrupts circadian rhythms and inhibits RORA expression. Restoring RORA activity or using RORA agonists can re-establish the cellular circadian clock [
41]. Given the typical circadian rhythmicity of hair follicle development and the pivotal role of RORA in circadian regulation, studies have focused on the relationship between the two. In our preliminary research, we found that RORA may also be involved in regulating the autophagy levels of hair follicle stem cells. Therefore, this study uses rat hair follicle stem cells as a model to analyze the impact of RORA on their autophagy levels and attempts to characterize the underlying molecular regulatory mechanisms.
2. Materials and Methods
2.1. Cell and Drug Treatment
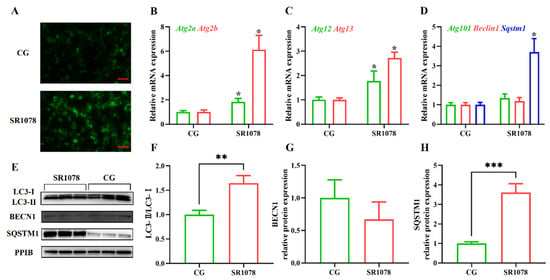
The cell model used in this study was primary hair follicle stem cells isolated from rat whiskers and long-term preserved in the laboratory. After 24 h of cell attachment and equilibration, the experimental group was treated with the RORA agonist SR1078 (MCE, Monmouth Junction, NJ, USA, HY-14422) at a final concentration of 10 μM, with a DMSO (1‰) control group set up. Chloroquine at 5 mM (MCE, HY-17589A) was used to block autophagy in cells to detect autophagic flux.
2.2. RNA Purification and Reverse Transcription
After treating the cells according to the experimental groups, the medium was removed, and the cells were washed three times with PBS buffer, with the supernatant removed as much as possible. RNA extraction from the cells was performed using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, Nanjing, China, RC112-01). The Qubit 4 Fluorometer and Qubit™ RNA IQ Assay Kit (Invitrogen™, Carlsbad, CA, USA, Q33221) were used to assess RNA integrity. The PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Kyoto, Japan, RR047A) was used for reverse transcription to obtain cDNA. The steps and protocols for RNA extraction, quality assessment, and reverse transcription followed the instructions provided with the kits.
2.3. Quantitative Real-Time PCR (qRT-PCR)
The obtained cDNA samples were analyzed by qRT-PCR using the SsoAdvanced™ Universal SYBR
® Green (Bio-Rad, Hercules, CA, USA, 1725270) and the Bio-Rad CFX384 Real-Time PCR Detection System to detect the relative expression levels of the genes. The reaction system and program settings followed the instructions provided with the kit. Melting curve analysis was performed using the default program of the instrument. The Cyclophilin B (
Ppib) gene was used as an internal reference gene for normalization of the results, and the 2
−ΔΔCt method was used for statistical analysis of gene differential expression levels. The primer sequences are provided in the
Supplementary Materials.
2.4. Total Protein and Nuclear Protein Isolation
After drug treatment, the medium was removed from the cells, and 0.25% trypsin containing EDTA (Gibco, San Francisco, CA, USA, 25200056) preheated to 37 °C was added to digest the cells. Digestion was terminated when the cell connections became loose, and the cytoplasm contracted by adding a complete medium containing serum. The cell suspension was transferred to a centrifuge tube and centrifuged at 200 g for 5 min, with the supernatant removed. The cell precipitate was resuspended in PBS buffer and centrifuged again to thoroughly wash away any remaining liquid. RIPA lysis buffer containing 1 mM PMSF was added to the cell precipitate, and the mixture was pipetted and vortexed. After thorough mixing on a vortex mixer, the mixture was transferred to ice and left to stand for 10 min, with vortexing performed multiple times during this period to ensure complete cell lysis. After centrifugation again, the supernatant was collected and transferred to a new centrifuge tube as the total cellular protein. Nuclear protein extraction was performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, Waltham, MA, USA, 78833), with experimental steps following the instructions provided with the kit.
2.5. Western Blot
Protein samples were quantified using the BCA method, with the loading volumes adjusted to be consistent. Subsequently, 4× Protein SDS-PAGE Loading Buffer (Takara, Kyoto, Japan, 9173) was added in proportion. The samples were treated at 99 °C for 10 min to ensure complete denaturation of the protein samples. Electrophoresis was performed using a 4–20% gradient SDS-PAGE gel (Genscript, Piscataway, NJ, USA, M00655) to thoroughly separate the protein samples. After electrophoresis, the protein bands were transferred onto a PVDF membrane at 200 mA for 1 h. The PVDF membrane was then blocked with 5% BSA (in TBST) for 1 h to prevent non-specific binding. Following blocking, the membrane was incubated overnight at 4 °C with primary antibodies (Proteintech, Rosemont, IL, USA, 14600-1-AP, 11306-1-AP, 18420-1-AP, and 11607-1-AP. The dilution concentrations are 1:3000, 1:5000, 1:15,000, and 1:3000, respectively). The next day, after removing the primary antibodies, the PVDF membrane was thoroughly washed with TBST buffer three times for 10 min each. The membrane was then incubated with secondary antibodies (Proteintech SA00001-2. The dilution concentration is 1:5000.) for 1 h at room temperature. After another thorough washing with TBST buffer, the proteins were visualized using the ECL method (Meilunbio, Dalian, China, MA0186-1). Image acquisition was performed using the Bio-Rad ChemiDoc MP Imaging System, and grayscale analysis of the bands was conducted using ImageJ software v1.8.0.
2.6. Immunofluorescence
Cells were washed three times with PBS buffer for 3 min each. They were then fixed with 4% paraformaldehyde for 15 min. After removing the fixative, the cells were washed again three times with PBS buffer. The cells were permeabilized with 0.5% Triton X-100 for 20 min at room temperature, followed by three additional washes with PBS. The cells were then blocked with goat serum for 30 min at room temperature. Primary antibodies were added (SQSTM1 dilution is 1:750; BECN1 dilution is 1:200), and the cells were incubated overnight at 4 °C. The next day, after removing the primary antibodies, the cells were washed three times with PBS for 3 min each. Secondary antibodies were incubated for 1 h at room temperature, followed by washing with PBS buffer. The cells were mounted with an anti-fluorescence quenching mounting medium containing DAPI and observed under a fluorescence microscope for image acquisition.
2.7. MDC Detection
Cell autophagy levels were detected using the cell autophagy staining detection kit with MDC (monodansylcadaverine) (Beyotime, Shanghai, China, C3018S), following the instructions provided in the kit. Briefly, after removing the cell culture medium from different experimental groups, the cells were washed with PBS buffer. One milliliter of MDC staining solution was added, and the cells were incubated at 37 °C in the dark for 30 min. The MDC staining solution was removed, and the cells were washed three times with Assay Buffer. One milliliter of Assay Buffer was added, and the cells were observed under a fluorescence microscope.
2.8. Cleavage Under Targets and Release Using Nuclease
The Cleavage Under Targets and Release Using Nuclease (CUT&RUN) technique was employed to enrich downstream target gene fragments bound by RORA. The experiment was conducted using the CUT&RUN Assay Kit (Cell Signaling Technology, Danvers, MA, USA, #86652) and DNA Purification Kit (Cell Signaling Technology, #14209), with an IgG control group set up. The steps followed the instructions provided in the kits. The enriched DNA samples from the RORA target gene binding regions were then subjected to droplet digital PCR for detection of the Sqstm1 promoter region.
2.9. Droplet Digital PCR
After appropriate dilution, samples were subjected to ddPCR using EvaGreen Digital PCR Supermix (Bio-Rad, CA, USA, #1864034). The reaction system and protocol were as specified in the kit’s instructions. A brief outline of the experimental procedure is as follows: the prepared PCR reaction mixture (20 μL) was transferred to a droplet generation card, and droplet generation oil (70 μL; Bio-Rad, #1864005) was added to the card. Water-in-oil droplets were generated using a droplet generator (Bio-Rad, #1864002) and transferred to a 96-well plate for PCR amplification. After the PCR reaction, droplets were read using a droplet reader (Bio-Rad, #1864003), and the experimental results were analyzed using Bio-Rad QuantaSoft™ Analysis Pro (QuantaSoft AP) softwareversion 1.4. Samples with more than 10,000 valid droplets analyzed were considered qualified for analysis.
2.10. Super-Shift Electrophoretic Mobility Shift Assay
Firstly, the motif structure of RORA-binding target genes was obtained from the JASPAR database, and a common specific sequence within these motifs was identified. This sequence was considered as the conserved region recognized and bound by the DNA-binding domain of the RORA protein. Probes targeting this region were designed and subjected to appropriate chemical modifications. Both wild-type and mutant probes were labeled with biotin, while cold probes (identical in sequence to the wild-type probes) were unlabeled. Electrophoretic Mobility Shift Assay (EMSA) analysis was conducted using the LightShift Chemiluminescent EMSA Kit (Thermo Fisher, 20148). A 6.5% native polyacrylamide gel was used for the separation of complexes, and a 0.45 μm pore size nylon membrane was used for blot transfer. The bands were visualized using the ECL method, and images were captured using the Bio-Rad ChemiDoc MP Imaging System. Steps followed the instructions provided in the kit.
2.11. Statistics
Each experiment included at least three biological replicates, and Student’s t-tests were used to assess statistical significance. All statistical analyses were performed using GraphPad Prism 9.5.1 software, and the results were expressed as mean ± standard deviation. p < 0.05 was considered to indicate a statistically significant result.
4. Discussion
The periodic reconstruction of hair follicles in mammals is a typical phenomenon of organisms’ response to environmental stress [
43]. Research on the developmental regulation of hair coat has been conducted for a long time, but progress has been slow, and we still know little about it. In particular, the inducing factors of hair follicle development or the transition between developmental stages deserve high attention. The factors regulating the periodic transition of HFSCs between activation and quiescence have been one of the research hotspots in this field in recent years. Autophagy, a conserved intrinsic homeostasis maintenance mechanism in organisms, is crucial in clearing aged and damaged proteins and organelles. Currently, the academic community is increasingly recognizing that autophagy may play a key regulatory role in hair follicle development. Numerous studies have analyzed the relationship between autophagy and hair follicle development, as well as the state of HFSCs [
44,
45,
46]. Research shows that when photobiomodulation therapy is used to promote hair growth, enhanced autophagy levels during the anagen phase of hair follicles are one of the key factors promoting their regeneration [
47]. Sun et al. found that induced autophagy promotes hair follicle development while blocking autophagy with 3-methyladenine leads to prolonged telogen and delayed anagen of hair follicles [
16]. Meanwhile, exogenous activation of autophagy can initiate the anagen phase of mouse hair follicles and promote hair growth [
6]. In addition, hair follicle development is regulated by multiple signaling pathways. Cai et al. found that the BMP and PTEN signaling pathways involved in hair follicle development regulate HFSC differentiation by modulating their autophagy levels [
48]. Studies on the regulation of hair follicle development by Myristoleic acid (MA) have shown that MA promotes growth hormone signaling by activating the Wnt/β-catenin and ERK pathways in dermal papilla cells and regulating autophagy and cell cycle progression [
49].
The
Sqstm1 gene plays a crucial role in various biological processes due to its complex domain structure. The SQSTM1 protein mediates phase separation through the polymerization of its N-terminal PB1 domain and the binding of its C-terminal UBA domain to ubiquitin [
50]. In addition, the PB1 domain at the N-terminus of SQSTM1 may interact with Rpt1 and S5a/Rpn10 of the 26S proteasome, while the C-terminal UBA domain binds ubiquitinated proteins, facilitating the degradation of short-lived, misfolded, and damaged proteins through the ubiquitin–proteasome system. The SQSTM1 protein also undergoes continuous and rapid nucleocytoplasmic shuttling via its two nuclear localization signals (NLS1 and NLS2) and a nuclear export sequence (NES), enabling the transport of ubiquitinated cargo between the nucleus and cytoplasm [
51,
52]. In terms of oxidative stress regulation, the SQSTM1 protein modulates cellular oxidative stress by influencing the NRF2-KEAP1 signaling pathway [
53,
54,
55,
56]. Most importantly, the SQSTM1 protein is highly involved in selective autophagy [
57]. The SQSTM1 protein binds to ubiquitin oligomeric chains of ubiquitinated substrates through its C-terminal UBA domain, while the PB1 domain assists in the packaging of ubiquitinated substrates through self-oligomerization. The LIR domain binds to the LC3B protein located on the isolation membrane, the precursor of autophagosomes, aiding the entry of ubiquitinated substrates into autophagosomes. Autophagosomes then fuse with lysosomes to form autolysosomes, ultimately completing the degradation of substrates [
58]. Our research results indicate that activated RORA binds to the promoter region of the
Sqstm1 gene to regulate its transcription level. The upregulation of SQSTM1 protein expression suggests enhanced interactions between SQSTM1 adapter proteins, ubiquitinated substrates, and LC3B protein on the isolation membrane, thereby increasing the efficiency of ubiquitinated substrate entry into autophagosomes and ultimately upregulating the level of selective autophagy (
Figure 4). The association between RORA and cellular autophagy has been demonstrated in related studies. During myocardial ischemia/reperfusion injury, cellular autophagy levels are significantly inhibited, and the absence of RORA significantly exacerbates this autophagy inhibition, suggesting that endogenous RORA may help restore autophagy function after myocardial ischemia/reperfusion injury [
59]. Research in cancer also shows that RORA significantly promotes autophagy levels in cancer cells [
60]. These findings corroborate the conclusion of our study that RORA promotes autophagy levels in HFSCs, but in previous studies focusing on the relationship between RORA and autophagy, the level of SQSTM1 protein was often considered to negatively correlate with the level of autophagy. However, our research has revealed that
Sqstm1 is a direct downstream target gene of RORA. Activation of RORA results in a significant upregulation of SQSTM1 levels in the hair follicle stem cells of rats. Meanwhile, changes in the LC3 II/LC3 I ratio and results from MDC staining all suggest an increase in autophagy levels. Therefore, we propose that RORA affects the autophagy level of HFSCs by upregulating selective autophagy.
RORA is recognized as a nuclear receptor for melatonin, which is an important molecule that transduces photoperiodic changes and has a well-established regulatory effect on the development of mammalian hair coats. This is also one of the important reasons why we focus on RORA [
61,
62,
63,
64,
65,
66]. Melatonin plays a crucial role in regulating biological rhythms as a bridge connecting photoperiodic signals and various physiological effects. The development of mammalian hair follicles exhibits obvious rhythmicity, further highlighting the importance of melatonin in hair follicle development. As the nuclear receptor for melatonin, RORA plays a vital role in mediating the physiological regulatory effects of melatonin. Studies have also shown that melatonin can regulate cellular autophagy levels through RORA [
59,
67,
68]. Given the regulatory relationship between autophagy levels and the state of HFSCs and hair follicle development, we speculate that melatonin can regulate the autophagy levels of HFSCs through RORA, thereby affecting hair follicle development. In addition, as a transcription factor highly involved in the regulation of various cellular physiological states, RORA has the potential to become a new research target in studies related to hair follicle development. More research can be conducted on the regulatory effect of RORA on the autophagy levels of HFSCs, aiming to provide a theoretical basis for the molecular regulatory mechanisms of hair follicle development and offer new insights into research on the treatment of diseases such as alopecia.