1. Introduction
Vascular smooth muscle cells (SMCs) account for the majority of cells in the walls of arterial blood vessels [
1,
2]. While the importance of vascular SMCs for the regulation of resistance arteries is accepted, their relevance in elastic arteries (e.g., aorta) is still underestimated [
3,
4]. Their contraction and relaxation are precisely regulated by different pathways to adapt vessel diameter and consequently fine-tune blood pressure [
5]. The second messenger molecule cyclic guanosine monophosphate (cGMP) is a key player in SMC relaxation [
6,
7]. Intracellular cGMP production is induced in two different ways. cGMP production by the cytosolic receptor soluble guanylyl cyclase (sGC) in response to nitric oxide (NO) is commonly known [
8,
9,
10]. In addition, the membrane-bound guanylyl cyclases-A (GC-A) and -B (GC-B) generate cGMP when binding their specific ligands, the natriuretic peptides (NPs) [
11]. ANP and BNP bind to GC-A while CNP activates GC-B. A third NP receptor, the NP clearance receptor (NPRC), binds the NPs with comparable affinity [
12]. The NPRC is thought to be mainly responsible for NP clearance; however, additional functions are suggested [
13,
14].
With cGMP being a key mediator of vasodilation, this signaling cascade is targeted by several cardiovascular therapeutics. While nitrates, sGC stimulators, and activators enhance sGC-dependent cGMP generation, phosphodiesterase inhibitors interfere with its degradation. NP-dependent signals are currently explored in cardiovascular disease. The BNP precursor NT-pro-BNP is an established biomarker for heart failure and NP-based therapeutics are being developed. The availability of NPs to bind to their signaling receptors GC-A and GC-B underlies additional local control. While NPRC competes for the NP ligand, the metalloproteinase neprilysin (NEP) degrades NPs. The latter one is also targeted by the NEP inhibitor sacubitril in the treatment of heart failure. In view of the different options to influence cGMP signals, it is of utmost importance to understand the intricate interplay of NPs with the different signaling components that shape the cGMP signal and illustrates an urgent need for establishing reliable models.
Vascular SMCs have been and are still intensively studied in cell culture experiments. Although cell culture essentially contributes to the basic understanding of intracellular signaling, it has been shown that in vitro experiments reflect physiological circumstances to a limited extent, and we hypothesize that the NP/cGMP cascade is no exception. Yet, systematic investigations comparing the cGMP system in intact tissue and corresponding cells are still scarce. In a previous study [
15], we evaluated the suitability of reference/housekeeping genes (RGs) for the comparison between intact aortic media and derived primary aortic SMCs by quantitative PCR (RT-qPCR). Besides the identification of U2 as a valid RG, we found that most RGs, especially classic examples such as beta-actin, did not fulfill the criteria for this comparison and showed a significantly different expression between the cells and the tissue. In our present study, we now aim to evaluate to what extent the expression of the cGMP signaling cascade changes when aortic SMCs are isolated and cultured.
The interest in the development of sex-specific medicine is increasing [
16]. This inevitably requires researchers to include the biological sex as a limiting factor for their studies. It has been shown that sex hormones can have a substantial impact on cardiovascular physiology [
17,
18,
19]. However, knowledge of the cardiovascular actions of NPs and their therapeutic potential in both sexes is mostly focused on the heart, while there is little information on aortic tissue [
20,
21]. Since we hypothesize that the NP/cGMP cascade differs between both sexes, we decided to additionally include a male and a female study cohort to explore sex-related differences in the cGMP signaling cascade.
2. Materials and Methods
2.1. Animals
Tissue was obtained from 2- to 3-month-old male and female Wistar rats (Rattus norvegicus) housed in the animal facility of the Veterinary Faculty at Justus-Liebig-University (JLU) Giessen, Germany. All the procedures were conducted according to the guidelines of the German Animal Welfare Act and approved by the Committee for Laboratory Animals of the JLU Giessen, case number JLU Nr. 577_M (approved period: January 2020–December 2022). The rats were anesthetized with CO2 and sacrificed by cervical dislocation.
2.2. Tissue Preparation
Aortae were dissected, transferred into MEM 1X medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 1% (v/v) Penicillin–Streptomycin (Pen/Strep, 10,000 U/mL, Sigma Aldrich, St. Louis, MO, USA), and stored on ice. The SMC layer (media) was prepared in cold MEM 1× + Pen/Strep. After removing the outer fat, branching vessels were cut, and the media was opened longitudinally from the aortic arch until the thoracic bifurcation. Onehalf of the same aorta was used for primary SMC extraction (see following chapter), while the second half was prepared as follows: Starting from the proximal end, the surrounding connective tissue (adventitia) was manually removed using curved tweezers. The endothelium (intima) was carefully scratched away. For RNA isolation, the remaining moisture was dried off the tissue, which was subsequently scaled, and immediately frozen in liquid nitrogen.
2.3. SMC Extraction and—Culture
The extraction of the SMCs from the aortic media required a two-step enzymatic digestion with collagenase (Collagenase Type II, Sigma-Aldrich, St. Louis, MI, USA). The aorta was prepared as described above and the longitudinal half designated for SMC extraction was first digested with collagenase (10% (w/v) in MEM 1× + Pen/Strep) at 37 °C for 15 min. After the transfer into fresh MEM 1× + Pen/Strep, the tissue was manually separated from the adventitia starting at the proximal end. The thin intimal layer was scratched off and the remaining media was cut into small pieces using sterile scissors. The media pieces were transferred into fresh MEM 1× + Pen/Strep with collagenase (10% (w/v)) and incubated for 30 min at 37 °C. Afterward, digestion was stopped by dilution with approximately 5 mL prewarmed cell culture medium DMEM/F-12 (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) + Pen/Strep containing 10% (v/v) fetal bovine serum (FBS, by Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The suspension was strained through a cell strainer (40 µM grid size by BD Biosciences, Bedford, MA, USA) under sterile conditions, and the flow-through, containing the singularized SMCs, was centrifuged for 10 min at 1500 rpm and room temperature. The cell pellet was resuspended in 5 mL fresh pre-warmed DMEM/F-12 + FBS + Pen-Strep and transferred into 25 cm2 sterile plastic cell culture flasks (Thermo Fisher Scientific, Waltham, MA, USA). The SMCs were cultured at 37 °C, 5% (v/v) CO2 and media was changed every 2 to 3 days. The extraction passage was cultured for 7 days before the SMCs were trypsinized (Trypsin-EDTA solution, Sigma-Aldrich) for 5 min at 37 °C after washing (DPBS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and transferred into fresh culture flasks. Cells (P1 till P3 after extraction) were seeded as follows: 4 to 5 × 105 cells per 25 cm2 flask for the RNA extraction and follow-up RT-qPCR, and 4 × 104 cells per well in 48-well plates for the cGMP ELISA.
2.4. RNA Extraction
Total RNAs from the isolated and cultured SMCs and from the intact media of the same animal were prepared using the RNeasy® Mini Kit (Qiagen, Hilden, Germany). Isolation was performed according to the manufacturer’s protocols using the spin technology with on-column DNase digestion (RNase-Free DNase Set (50), Qiagen, Hilden, Germany). For isolation from intact tissue, approximately 10 to 20 mg of rat aortae (male and female, each n = 12) were frozen in liquid nitrogen and pulverized using a hammer. RLT buffer with 1% (v/v) 2-Mercaptoethanol (Roth, Karlsruhe, Germany) and a sterile metal bead were added, and the sample was processed for 3 min at 300 Hz in a tissue lyser mixer mill MM400 (Retsch, Haan, Germany). The lysate was centrifuged (5 min at 12,000 rpm) and the supernatant was further processed according to the manufacturer’s instructions. The final RNA concentration was determined using a NanoDropTM 2000/2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The RNA samples were stored at −80 °C.
2.5. cDNA Synthesis
A total of 2 µg of the extracted RNA served as the template for the cDNA synthesis. SuperScriptTM II Reverse Transcriptase and Oligo(dT)20 primer (both from Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) were applied according to the manufacturer’s instructions using the Mastercycler gradient 5331 (Eppendorf, Hamburg, Germany). The consistency of the reverse transcription (RT) was verified by the measurement of the cDNA concentration. In case of large differences, cDNAs were diluted to the same concentration for the subsequent RT-qPCR measurement.
2.6. RT-qPCR
RT-qPCR was performed using the iCycler IQ PCR system and the iQTM SYBR
® Green Supermix (both from Bio-Rad, Hercules, CA, USA) using the cycler steps listed in
Table 1 below. For the quality control of the RT-qPCR, primers and -RT controls were applied right next to the PCR products on a 3% agarose (
w/
v) ethidium bromide gel). The length of the double-stranded PCR products was checked after visualization in a UV chamber (Gel Jet Imager running the software Intas GDS Touch 2, Intas Science, Göttingen, Germany).
Custom DNA-Oligos (0.01 µM, salt-free, lyophilized, MALDI-TOF MS quality analysis) were purchased from Eurofins, Hamburg, Germany. Primer details for the RG and the genes of interest are listed below. The efficiency (
E) of each primer pair was calculated based on a cDNA dilution series with three steps (undiluted, 1:2, and 1:4). Every dilution sample was measured as a triplet in a usual RT-qPCR run according to the qPCR steps given in
Table 1. The resulting Ct mean values of each dilution level were plotted against the concentration of the diluted sample (log10 scale) and used to create a descending slope (
m) by linear regression according to the following calculation:
Under the ideal conditions of the PCR and 100% efficiency of the primers, a duplication of the nucleic template sequence can be expected. We only used primer sets that achieved an efficiency between 100–120%.
Small nuclear RNA—U2:
Forward 5′-ATCTGATACGTCCTCTATCC-3′
Reverse 5′-GTGGACGGAGCAAGCTCCTA-3′
Amplicon size: 83 bp
Efficiency: 102%
Guanylyl cyclase A—GC-A:
Forward 5′-GACCTACTGCCCTGCTGTTC-3′
Reverse 5′-GAGAGGTGGAAGCTGGACAC-3′
Amplicon size: 77 bp
Efficiency: 102%
Guanylyl cyclase B—GC-B:
Forward 5′-TGGCTCTTAGGAGAGCGAAA-3′
Reverse 5′-GCCCATCCAAGTACCAACAG-3′
Amplicon size: 84 bp
Efficiency: 119%
Natriuretic peptide clearance receptor—NPRC:
Forward 5′-CTCCTTGCAAATCATGTGGCCT-3′
Reverse 5′-TCTTGGTGATTTCGCCTCTCA-3′
Amplicon size: 142 bp
Efficiency: 120%
Soluble Guanylyl cyclase subunit beta 1—sGC:
Forward 5′-GCGGACACCATGTACGGTT-3′
Reverse 5′-GCAGCCACCAAGTCATAGGT-3′
Amplicon size: 170 bp
Efficiency: 115%
The relative mRNA amount of the target genes was calculated using the ΔCt method by normalization to the RG U2 [
15].
2.7. cGMP ELISA
cGMP levels after the NP treatments were determined by ELISA (Institute for Hormone and Fertility Research [IHF], Hamburg, Germany) based on a previously described assay [
24,
25] following the principles of a competitive ELISA. Therefore, Immuno 96-well plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated with goat-anti-rabbit IgG (H + L) antibody solution (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Sodium chloride, Tween, Hydrogen peroxide (H
2O
2), and Tetramethylbenzidine (TMB) were obtained from Roth. Sodium acetate was obtained from Merck, Darmstadt, Germany, and citric acid from Sigma-Aldrich. cGMP standard (Biolog, Hayward, CA, USA) dilution series was prepared in E-PBS [0.1 M sodium triphosphate, 0.15 M NaCl, 5 mM EDTA, 0.2% (
w/
v) BSA, 0.01% (
w/
v) Thimerosal; (pH 7.0)].
For the cGMP ELISA, the SMCs were cultured in 48-well plates until 80% of the well bottom was covered. The cell culture media was exchanged with FBS-free MEM 1× + Pen-Strep for at least 4 h for equilibration. For treatment, MEM 1× + Pen-Strep was prewarmed and 3-Isobutyl-1-methylxanthine (IBMX, 0.25 nM, Sigma-Aldrich, St. Louis, MI, USA), a phosphodiesterase inhibitor dissolved in Dimethyl sulfoxide (DMSO, Roth, Karlsruhe, Germany), and ANP or CNP (each 100 nM, Bachem, Bubendorf, Switzerland) were added. The same medium with IBXM only was applied as the control medium. Treatment at 37 °C and 5% (v/v) CO2 was stopped after 30 min by the addition of ice-cold 100% (v/v) ethanol and immediately freeze down at −20 °C for at least 1 h. The cells and supernatant were detached and centrifuged for 30 min at 4 °C and 20,000× g. Clear supernatant was transferred into a fresh tube and lyophilized. The remaining pellet was resuspended in 140 µL E-PBS.
Longitudinal halves of one aorta, prepared as mentioned above (see Tissue preparation), were scaled and equilibrated in prewarmed MEM 1× + Pen-Strep for 10 min at 37 °C. For treatment, the tissues were incubated with MEM 1× + Pen-Strep containing IBMX (0.25 nM) and ANP or CNP, each at a concentration of 100 nM for 30 min at 37 °C. One longitudinal half was treated with ANP, and the other half of the same vessel was treated with CNP. The supernatant was frozen immediately in liquid nitrogen and stored at −80 °C until measurement as follows.
The samples and standards (50 µL per well) were loaded in duplicates on the antibody-coated 96-well plates together with 100 µL cGMP antiserum [1:80,000] and 50 µL Biotin-cGMP tracer [170 fM] were added according to manufacturer’s suggestions. The plates were incubated for at least 18 h in a dark, wet chamber at 4 °C. The solution was removed from wells and 200 µL of HPR-Streptavidin solution [0.152 µg/mL in E-PBS] (Vector Laboratories, San Francisco Bay Area, CA, USA) was added per well. The plate was incubated for 30 min in a dark, wet chamber at 4 °C. The wells were washed 3 times with washing buffer [0.5% NaCl (w/v); 0.02% Tween 20 (v/v)], and 250 µL HPR-substrate solution [0.01 M sodium acetate; 0.0045 M citric acid; 0.0038% H2O2 (v/v) and 0.001% TMB (w/v)] was added per well. After an incubation for 40 min in a dark, wet chamber at room temperature, 50 µL H2SO4 2M (Roth) was added. The emission intensity was detected photometrically by a DIAS microplate reader (Dynatech Laboratories, Denkendorf, Germany) at 450 nm. Based on the blanks and the standard, the concentration of cGMP in the samples was calculated automatically by the software Revelation 2.0 (Dynatech Laboratories). For the tissue, additionally, the cGMP concentration was normalized to the tissue weight.
2.8. Statistical Analysis
For all the underlying statistical analyses, GraphPad Prism version 10.2.2 (397) for Windows (GraphPad Software, La Jolla, CA, USA,
www.graphpad.com, last accessed for analysis: 15 April 2024) was used. All data were pre-checked for normal distribution. The n-number (given in each figure) refers to the number of biological replicates (number of animals). Detailed descriptions of the statistical evaluation and significant differences are given in the corresponding figure legends. Data are depicted as the median ± interquartile range.
4. Discussion
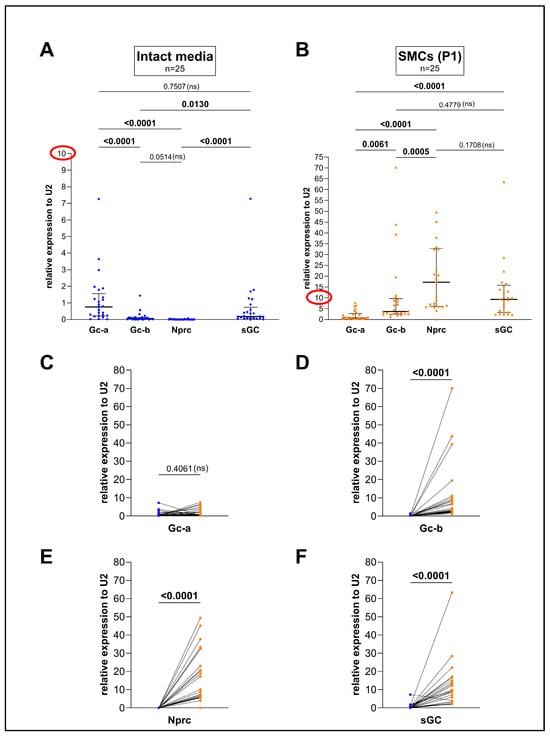
The investigated genes GC-B, NPRC, and sGC showed a dramatic increase in transcription in the cultured primary SMCs when compared to the corresponding intact aortic media (see
Figure 1). The mRNA level of GC-A, however, remained stable throughout cell extraction and culturing.
In recent decades, isolated and cultured SMCs have been used regularly to characterize the expression and function of SMCs in intact tissues including vasculature. The stable expression of
Gc-a was in contrast to early studies by Suga and colleagues [
26,
27]. They suggested a downregulation of
Gc-a expression in primary rat SMC culture. Their conclusions mostly relied on Northern blot analysis using the common housekeeping gene beta-actin. We previously tested several RGs by qPCR [
15], amongst them, classical housekeeping genes such as beta-actin, and found that the expression of beta-actin differed dramatically between the intact aortic media and cultured primary aortic SMCs. The difference in the
Gc-a expression between Suga et al. and our study could be due to the use of beta-actin as RG. When using the RG U2 [
22] previously validated for the comparison of intact aortic tissue with its corresponding primary SMCs [
15], it was surprising to see that
Gc-b but not
Gc-a expression significantly changed upon culturing. Since both receptors not only share their localization within SMCs [
28,
29] but also structural and regulatory features [
11,
29,
30], a differing role of both cGMP-producing NPRs in cultured SMCs in comparison to intact tissue can be assumed, most likely due to a switch in the SMC phenotype [
31,
32]. Furthermore, it could be concluded that
Gc-a, with its stable expression, is the only gene investigated that was found at the “physiological” (tissue) level. It should be mentioned that this does not necessarily imply that GC-A fulfills the same functions in the cultured cells as it does in the intact tissue.
However, when defining an RG, a gene expression unimpaired by experimental conditions is crucial [
33]. Regardless of its biological function, this could indicate the suitability of
Gc-a as a potential housekeeping gene for the comparison of intact vascular tissue and cultured SMCs.
The gene that stood out by the most impressive increase in expression upon culturing of all the genes investigated in this study was the NP clearance receptor Nprc [
12]. The reasons for the increase in
Nprc expression remain speculative. First and foremost, it is likely that an increase in NP clearance counteracts the excessive activity of cGMP signaling by NPs in cell culture. The simultaneous increase in
Gc-b (and
sGC), however, suggests further NPRC functions in cell culture. It has been reported earlier that the functions of NPRC reach beyond that of the mere NP clearance [
13,
14,
34]. One function could be the cellular response to oxidative stress which was found to be attenuated by enhanced NPRC expression [
35]. In addition, the anti-proliferative capacities of NPRC were suggested for vascular SMCs [
36]; an increase in
Nprc expression, thus, could be the response to a boost in proliferation or an increased oxidative stress, both evident in cultured vascular SMCs [
37]. Since the increase in
Nprc expression seems to happen upon culturing,
Nprc may be considered as a potential marker gene to identify the synthetic phenotype of vascular SMCs.
Besides the receptors for the NPs, we decided to additionally investigate the expression of
sGC, the receptor for NO-induced vascular SMC relaxation [
9,
10,
38]. It is of interest that the cytosolic cGMP-producing
sGC also shows an increased expression (see
Figure 1F) like the NP receptors
Gc-b and
Nprc upon the culturing of the vascular SMCs.
Awareness of sex differences become increasingly recognized in the field of cardiovascular research. Male and female patients show different symptoms associated with certain diseases [
39,
40,
41] or show different responses to treatment and medication [
16,
42]. Also, in vitro studies have shown that the two sex hormones (testosterone versus estrogen) may influence cultured vascular SMCs in different ways [
18,
42,
43]. This increasing awareness regarding the potential influence of the biological sex led us to include an equal number of both male and female study subjects to investigate the NP/cGMP cascade.
When comparing the male and female cohort, their expression profiles seemed very similar (see
Figure 2G,H); however, a few differences in the sexes were noticeable. Only in the female primary SMCs,
Nprc significantly exceeded the expression of
Gc-b and
sGC (see
Figure 2C,D) correlating to a much higher mean value of NPRC expression in the female cohort. However, when comparing
Nprc expression between males and females directly (see
Figure 2H), no significance was found most likely due to a larger spread in the female group. Such larger spreads in the female group were also found for other genes (see
Figure 2B,D). Since all the female rats included in this study were of the same strain, age, and nearly the same weight, this variability may be explained by different estrus stages. Thus, it would be most interesting to correlate the level of expression with the hormonal status, determined at the time of organ harvest [
44]. Previous studies in the heart have already shown sex-specific differences in ANP-induced cGMP [
20].
We determined the enzyme activity of GC-A and GC-B by cGMP ELISA upon ANP or CNP treatment in intact tissue and cultured SMCs. In intact aortic tissue, the cGMP measurements showed a significant difference between the ANP- and CNP-induced cGMP production in the male but not in the female study group (see
Figure 3B,C) after the treatment of one half of the aorta with ANP and the other with CNP and using a pairwise analysis. However, when comparing ANP-induced cGMP levels between male and female rats using an unpaired analysis, no significant difference was visible (see
Figure 3E). Comparable results were found for the CNP effects (see
Figure 3F). The significance in the pairwise analysis might be due to the large spread of the ANP-induced cGMP effects. The reasons for this spread in intact tissues, normalized to the tissue weight, are not yet understood. Similar significant effects were found in the cultured SMCs (first passage after extraction) when comparing sex-specific ANP- and CNP-induced cGMP production using pairwise analyses (see
Figure 4B,C) in contrast to ANP- or CNP-effects separately with non-pairwise analysis (see
Figure 4E,F).
In the SMCs of passage 1 (P1), ANP treatment induced higher cGMP levels than CNP (see
Figure 4A). This was inverse to the mRNA expression of the ANP and CNP receptors in the SMCs of the same passage and from the same animals, i.e.,
Gc-b significantly exceeded
Gc-a expression (see
Figure 1B). In the following passages (P2, P3), however, the CNP-induced cGMP production of the cultured cells began to exceed the ANP-induced cGMP production. Even though this effect was readily visible in graphs (
Figure 5), it did not reach statistical significance when performing pairwise comparisons. This might reflect that the typical
Gc-a and
Gc-b pattern at the mRNA level (
Gc-b mRNA >
Gc-a mRNA) in cell culture has now been translated to the protein level (GC-B > GC-A) visible by the receptors’ cGMP production.
Our data illustrate that NP receptor expression seems to rapidly adopt a new balance upon cell isolation with subsequent changes in receptor functionality. With the remarkable increase in Nprc expression, the local control of NP availability is affected and might change regular NP-induced cGMP signals. These disrupted signals in cell culture may obscure physiological mechanisms and lead to the misjudgment of the entire pathway impeding, e.g., drug discovery.
In this regard, it is interesting that cultured vascular SMCs share characteristics with SMCs in the walls of diseased blood vessels [
31,
45]. Thus, it would be interesting to see whether a similar expression profile is found in the tissue of aortic pathology compared to SMC culture. Cultured vascular SMCs may potentially serve as an in vitro disease model in the future [
2,
46].
Given that the isolation of cells rapidly induces changes at the transcription level, RNAseq data should be interpreted with caution. Emerging techniques like spatially resolved transcriptomics using intact tissue slices may provide a more realistic view of the protein expression profile and hold the potential to unveil new yet unknown clinically relevant biomarkers that may be missed in single cell-based approaches.