
1. Introduction
Box 1. Biological effect of DNA methylation.
significant role in cancer development. The altered DNA methylation patterns
affect various genes in our body and influence essential biological processes
such as genome stability, inflammation, cell cycle regulation, and metabolism
[33].
The changes typically involve two main processes: hypomethylation (a decrease
in methylation across the genome) and hypermethylation (an increase in
methylation in specific regions, such as CpG islands) causing cancer.
1. Hypomethylation
The loss of methylation in these regions heightens genomic instability by
activating transposable elements and causing insertional mutagenesis [34].
b. Centromeric and Telomeric Regions: Hypomethylation in these areas
contributes to chromosomal instability and is associated with shorter
telomeres related to aging and cancer [35].
hypomethylation can contribute to cancer development, a common age-related
disease [36].
may lead to increased APP expression, resulting in excessive amyloid-beta
(Aβ) production, which forms the plaque characteristic of Alzheimer’s disease
[37].
frequently observed in the promoter region of the SNCA gene in Parkinson’s
disease (PD). This hypomethylation increases α-synuclein expression,
contributing to its aggregation and the formation of Lewy bodies, a hallmark
of PD [38].
crucial for keeping pluripotency-associated genes, such as OCT4, SOX2, and
NANOG, active. During the differentiation process, there is a transition
towards hypomethylation in lineage-specific genes, allowing their activation [39].
Understanding the specific roles of hypermethylation and hypomethylation is
essential for enhancing stem cell-based therapies in regenerative
medicine.
2. Hypermethylation
of Cytokine Signaling 1) regulates cytokine signaling and inhibits
tumorigenic pathways, specifically the JAK-STAT signaling pathway, and is
frequently hypermethylated in cancers [40]. (b) CDKN2A
(p16, p14ARF) controls cell cycle progression; its silencing leads to
unchecked cell division and is associated with aging and cancer [41].
(c) PTEN is a phosphatase that plays a pivotal role in negatively regulating
the PI3K/AKT pathway, preventing uncontrolled cell growth [42].
(d) MLH1 is a crucial mismatch repair (MMR) gene, and its silencing results
in microsatellite instability (MSI) observed in cancers [43].
(e) TP53 regulates DNA repair processes, apoptosis, and cancer. TP53 is
rarely hypermethylated in cancers [44]. The nuclear
entry of p53 and autophagy in cancer cells are associated with a circular RNA
circ-Dnmt1, particularly in the epigenetic regulation of autophagy-related
genes [45].
diminish immune responses. Additionally, hyper-methylation of
anti-inflammatory or immune-regulatory genes can lead to dysregulated
production of pro-inflammatory cytokines, contributing to inflammation [46].
often exhibit hypermethylation in aging cells. The silencing of BRCA1, MLH1,
and MGMT diminishes DNA repair efficiency, resulting in genomic instability
and contributing to the aging process and cancer [47].
development: (a) Hypermethylation: RB1 regulates the cell cycle checkpoint [48].
(b) Hypomethylation: E2F targets transition from the G1 to the S phase [49].
region can suppress its expression, diminishing its role in stress response
and energy metabolism, which is related to metabolic disorders, chronic
inflammation, aging, and cancer development [50].
associated with decreased expression, which adversely affects mitochondrial
biogenesis and energy homeostasis, commonly observed in age-related metabolic
disorders and cancer [51,52].
3. Dysregulated methylation
hypermethylation can influence the expression of these pro-inflammatory
cytokines. Hypomethylation of promoter regions can lead to increased
expression, while hypermethylation of regulatory regions can result in reduced
expression. However, chronic low-grade inflammation associated with aging and
cancer is often linked to diminished regulation of these genes. This suggests
a dysregulation of methylation patterns, including hypermethylation of
regulatory regions and hypomethylation of promoters [53,54].
or in certain diseases, hypermethylation of HOX gene promoters can lead to
the silencing of these genes. This silencing disrupts normal cellular
differentiation and tissue maintenance, contributing to dysfunction
associated with aging or diseases like cancer. For example, hypermethylation
of specific HOX genes has been observed in tumors, resulting in abnormal
differentiation [55]. (b) Hypomethylation: Hypomethylation
in HOX genes’ regulatory regions can cause inappropriate reactivation or
misexpression [56]. This may result in the undesired
activation of developmental pathways in adult tissues, leading to
dysregulation that could contribute to cancer progression.
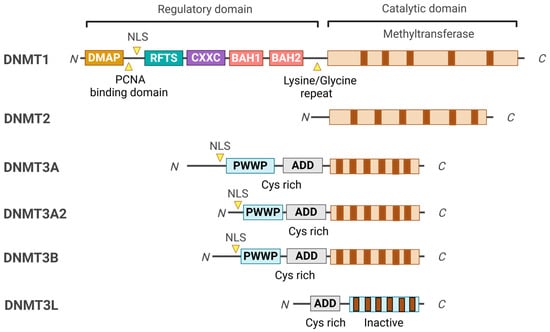
2. DNMT Domain Organizations and Their Functions
4. DNMT1 Binding Partners
- (1)
MBDs
- (2)
CFP1
- (3)
DMAP1 (DNMT-associated protein1)
- (4)
UHRF1
- (5)
Sp1 (Specificity protein 1)
- (6)
PCNA (Proliferating Cell Nuclear Antigen)
- (7)
G9a/GLP (Histone Methyltransferases)
- (8)
RUNX1-MTG8
- (9)
HESX1 (HESX homeobox 1)
- (10)
DAXX
- (11)
CHAF1A
5. DNMT1 Regulators
- (1)
MEK/ERK
- (2)
STAT3
- (3)
MicroRNAs
- (4)
Circular RNAs
6. DNMT1 Inhibitor
7. DNMT Family and Cancer
Hypomethylation by suppressing DNMT1 activity occurs throughout the genome, reducing 5mC, particularly in gene coding regions and satellite repeats.
8. Conclusions and Future Perspectives
Beyond tumors, DNMT inhibitors could play a role in restoring normal gene expression in neurodegenerative and psychiatric disorders. That could benefit regenerative therapies using embryonic stem cells and induced pluripotent stem cells (iPSCs). Future directions should focus on developing DNMT inhibitors that are less toxic and specifically target different DNMT isoforms and cancer cells. Combining promising novel DNMT1 inhibitors with other therapeutic strategies, such as chemotherapy, radiotherapy, vaccines, immune checkpoint inhibitors, and CAR-T therapies, may yield effective synergistic responses in cancer treatment.
Source link
Dae Joong Kim www.mdpi.com