1. Introduction
The advent of stem cell research and the generation of organoid models using human embryonic stem cells (hESC) or, in particular, human-induced pluripotent stem cells (hiPSCs) has seen personalised models of neurodevelopmental disorders made possible for the first time [
1,
2]
These human-derived stem cells multiply continuously in monolayer cultures while retaining the ability to differentiate into nearly any human cell type [
1,
2] The hiPSCs originate as mature somatic cells, commonly fibroblasts obtained from skin samples, and are converted into pluripotent stem cells using reprogramming factors [
1,
2,
3]. Several different approaches are available for differentiation into monolayer cultures, including dual-SMAD inhibition or the overexpression of transcription factors such as Neurogenin-2 [
4]. Similarly, specific differentiation protocols are used for the generation of 3D organoid models, enabling the creation of more complex structures.
Three-dimensional (3D) brain or cerebral organoids have emerged as a ground-breaking technology with the potential to combine traditional laboratory findings with personalised clinical treatments [
5,
6]. These self-organising 3D structures closely mirror the neuronal organisation and functional activity of the developing human brain [
2,
5,
6]. Depending on the protocol applied, they may represent cells from all brain regions, like cerebral organoids or the patterning of specific regions, such as the cortex, yielding so-called corticoids. The ability to produce cortical-specific organoids is crucial for investigating disease mechanisms of severe developmental disorders like developmental and epileptic encephalopathies (DEE) [
7,
8], autism spectrum [
9,
10] and Angelman syndrome [
11]. In addition to the pathophysiology of neurodevelopmental disorders, these models can be used for therapeutic screening in a tissue-relevant context.
Despite the promise of organoid technology, small changes made by individual laboratories when implementing differentiation protocols contribute to structural heterogeneity in organoid models. Previous efforts to standardise organoid differentiation have aimed to reduce variability [
12], but any adopted protocol still requires minor adjustments based on specific cell lines and procedures. Notably, the size, shape and viability of cortical organoids can vary significantly both within and between batches. To address this heterogeneity, we sought to create an image analysis workflow specifically designed to examine large datasets and benchmark the selected protocol. This imaging workflow would serve as an important exercise for the individual lab to achieve a level of standardisation. Specifically, it is to be applied to existing or newly adopted differentiation protocols to assure the consistency of data across experiments.
Evidence of cellular diversity in organoid models has primarily been drawn from measures of cell function, gene expression and protein abundance; however, these give little insight into architectural patterns of cell growth [
13,
14]. Recently, many groups have focused on high-throughput imaging methods to assess organoid morphology [
15,
16,
17,
18], and 3D spatial single cell and proteomic-based analysis methods have also emerged [
19]. However, at the cellular level, high-resolution imaging techniques are still needed to measure the integrity of maturing organoids, which is challenging when applied to cortical organoids [
13,
14]. Additionally, the need for large sample datasets to obtain statistically valid comparisons creates a bottleneck in the imaging process. The ability to classify and count many cells via an automated system offers a promising solution to accelerate tissue analysis. Automating this process is especially important, as these models become more structurally complex as they mature. Current methods for evaluating organoids’ intricate morphology using microscopy images include a variety of open source and commercially available tools (e.g., Fiji/Image J, CellProfiler and Imaris). These tools often require extensive tailoring to specific requirements.
Brain organoid maturation facilitates synaptic and network development, necessitating extended culturing periods, which ultimately results in larger tissue sizes [
20,
21]. Investigating the cellular architecture of these larger tissues often involves sectioning tissue and employing immuno-histochemistry (IHC), a widely used procedure for organoid analysis [
14]. Other methods, such as tissue clearing, two-photon imaging, and light-sheet imaging, require specialised expertise and imaging systems, making them less commonly used [
14,
19,
22]. Our objective, therefore, was to develop a workflow using tissue sections as a more widely applicable image analysis method that could benefit various research groups.
One major challenge associated with large avascular organoids is their susceptibility to developing a necrotic core. A comprehensive structural analysis method must include a measurement approach that filters out dying cells and the necrotic core region [
23]. The absence of a vascular system limits the diffusion of oxygen and nutrients from the surface of organoids, leading to the formation of a dead cell core as the organoids grow [
24,
25]. Despite the emergence of methods to improve nutrient diffusion, physical constraints limit nutrient flow to approximately 400 µm, and in long-term 3D organoid cultures, hypoxic core susceptibility remains an issue [
26,
27,
28]. Changes in the volume of dead cell cores or the percentage of dead cells may also reflect features of the disease model [
4,
29,
30]. Image-based analyses have traditionally relied on the manual delineation of non-viable tissue boundaries, a process that is subjective, time-consuming and impractical for large sample sizes.
Another critical measurement we sought to address was obtaining cell counts for proteins expressed outside the nucleus or in peri-nucleic regions. While robust co-localisation imaging methods with cell nuclei are available, antibody labelling without spatial overlap—labelling non-nuclear proteins—presents a significant analytical challenge. Assigning these non-nuclear structures to a specific cell is non-trivial, as illustrated by antibodies marking filamentous structures. For example, microtubule-associated protein (MAP-2) is expressed in neuronal perikarya and dendrites, which, in cerebral organoids, have complex boundaries and lack stereotypical and layer-specific projection patterns [
31,
32,
33]. Alternatively, mean fluorescence intensity is often used to measure overall antibody expression; however, this approach does not account for tissue size nor reveal the number of cells expressing specific markers [
22,
32,
34,
35]. This can lead to misinterpretation, as low cell density and high fluorescence signal might resemble high density and weak fluorescence signal when averaged over a large area. By enabling more precise cellular-level statistics, our approach to estimating cell numbers enhances research utilising organoid models of human disease.
Overall, our objectives were to enhance whole-organoid image analysis by providing a way to identify and separate areas of cell death in non-viable regions and quantify cell number and density of non-nucleic antibody markers in viable tissue areas. The resulting analysis workflow incorporated machine-learning-based cell nuclei segmentation implemented via Cellpose v2.2 (Howard Hughes Medical Institute, Ashburn, VA, USA) providing masked images that were further processed in MATLAB (Version R2022b, Mathworks, Natick, MA, USA) via a custom Graphical User Interface (GUI). The analysis workflow was developed to facilitate processing large datasets of tiled and z-series images via progression through a semi-automated step-sequence.
2. Materials and Methods
2.1. Ethics
Ethics for the project were approved by the Austin Health Human Research Ethics Committee (HREC/16/Austin/472, Melbourne, Australia), and this study was conducted in adherence to the established guidelines set forth by the Austin Hospital, University of Melbourne, and the National Health and Medical Research Council (NHMRC) of Australia.
2.2. Human iPSC Model
Organoids were derived from two control cell lines, a hiPSC line derived from a neonate (Cat #WC026i-5807-3, WiCell, Madison, WI, USA) and an embryonic stem cell (ESC) line (Cat #H9 or WA-09, WiCell, Madison, WI, USA). Three batches of cortical organoids were generated from each cell line using the protocol established by Velasco et al. [
12]. Each batch comprised 12 organoids at two developmental stages: 6 were collected on day 120 (4 months) post-induction and 6 on day 180 (6 months).
2.3. Immunohistochemistry
At 4 and 6 months post-induction, a minimum of two technical replicates were immersed in 4% paraformaldehyde and incubated for 2 h at 4 °C, and then washed three times in 1x phosphate-buffered saline (PBS), and placed in tissue-Tek O.C.T. Compound (Cat #25608-930); then, they were snap-frozen and stored at −80 °C. Serial cryo-sections were cut at 20 µm thickness (Cat #CM1950; Leica Microsystems, Wetzlar, Germany), collected on SuperfrostPlus glass slides (Cat #I6172PLUS, Thermo Fisher, Waltham, MA, USA) and stored at −80 °C.
Immunohistochemistry was performed according to Yakoub and Sadek [
36]. All primary and secondary antibodies are listed in
Table 1. Sections were defrosted for 15 min at room temperature (RT), and then blocked using 10% normal goat serum (NGS; Cat #16210064; Invitrogen, Carlsbad, CA, USA) and 0.3% Triton X-100 (Cat#T8787; Sigma-Aldrich, St. Louis, MO, USA) in PBS, for a minimum of 2 h, at RT. Primary antibodies were diluted in an antibody vehicle solution (AVS) of 1% NGS and 0.1% Triton X-100 in PBS), and sections were incubated overnight at 4 °C in the dark.
Table 2 details the number of sections included in our analysis. Slides were washed in a solution of 0.05% Tween20 (Cat#655204; Sigma-Aldrich, St. Louis, MO, USA) and PBS and incubated with secondary antibodies (diluted in AVS) for one hour, at RT (see
Table 1). A second wash using a solution of 0.05% Tween20 and PBS preceded a 5 min incubation with 6-diamidino-2-phenylindole (DAPI). Two final PBS washes were undertaken then sections were mounted using Invitrogen ProLong Gold Antifade mounting medium (Cat #P36934, Thermo Fisher, Waltham, MA, USA) and covered with Menzel–Glaser glass coverslips (Cat #11911998, #1.5H, Thermo Fisher, Waltham, MA, USA). Slides were subsequently stored at −80 °C until imaged.
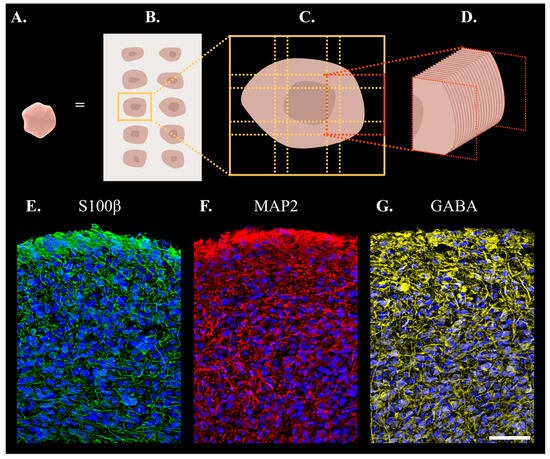
To characterise the morphology of the organoids with respect to protein markers indicative of mature expression patterning, three antibodies were used. In mammalian cells, S-100 (β-subunit) (S100β) protein is predominantly expressed by mature astrocytes and distributed throughout the cytoplasm [
37,
38,
39,
40]. Microtubule-Associated Protein 2 (MAP2) is a cytoskeletal protein mainly expressed in the soma and dendrites of mature neurons [
31]. Gamma-aminobutyric Acid (GABA)ergic neurons are inhibitory neurotransmitters chiefly located in the cytoplasm and dendrites of neurons [
41,
42]. A total of 88 sections were included in the final analysis, as outlined in
Table 2. Additional organoid sections (
n = 6) were included to verify non-viable cellular and core features using cleaved-caspase-3 antibody and TUNEL assay kit (
Table 1).
2.4. Imaging
Images of each organoid slice were acquired using a 20x/0.8NA objective on a Leica THUNDER Imager DMI8 fluorescence microscope (Leica Microsystems, Wetzlar, Germany). Image z-stacks with a z-step size of 0.571 µm and a pixel size of 0.323 µm xy were acquired to ensure adequate sampling in line with recommendations for post-processing using Leica Application Suite X (LASX, version 3.8.1.26810, Leica Microsystems, Germany). A tile scan function was used to rapidly image the entire organoid slice, with a 10% tile overlap. All sections were labelled with DAPI for nuclei segmentation (excitation wavelength used for imaging 391 nm) and either S100β (ex 479 nm), or MAP2 or GABA (ex 554 nm). The images were subject to small volume computational clearing (SVCC) using LASX software [
43] before being exported. Settings included feature scale—6000 nm and 30% background scale. In addition, confocal images were also acquired on a subset of organoid slices using an LSM900 scanning confocal microscope 20x/0.8NA (Carl Zeiss Inc., Jena, Germany). Confocal imaging time was considerably longer at approximately 6 h (parameters optimised for Nyquist sampling), versus 10 min on Leica THUNDER (parameters optimised for LASX processing as noted above). Only images acquired using the Leica THUNDER system were used for the analysis workflow development. The Leica THUNDER system presented a significant advantage to achieve high throughput and was used for analysis workflow development as it captured sufficient image quality to segment and analyse antibodies for cell counts. All images were acquired within the Florey Neuroscience Microscopy Facility (The Florey Institute of Neuroscience and Mental Health, Melbourne, VIC, Australia).
2.5. Image Data Analysis
A custom set of MATLAB (Version R2022b, Mathworks, Natick, MA, USA) functions were developed for the purpose of image analysis. These incorporated a range of steps, including tile stitching, nuclei and peri-nucleic antibody segmentation, cell-state characterisation, the identification of the non-viable cell core boundary and applicable calculations. A description of the image analysis workflow is incorporated in the results section outlined below. All analysis scripts are available on request. The analysis workflow was executed on a Dell PowerEdge R820 Server, equipped with four Intel Xeon E5-4620 processors (2.20 GHz, 16M Cache, 7.2GT/s QPI, Turbo, 8 Core) and supported by a RAM capacity of 512 GB.
2.6. Statistical Framework
All statistical analyses and figures relating to the visualisation of data were completed using GraphPad PRISM software (Version 9.5.1, GraphPad Software Inc., La Jolla, CA, USA). Numeric values are reported to three significant figures, and data are presented as mean ± standard error of the mean (SEM). To compare groups, multiple t-tests with Welch’s correction were conducted to account for unequal variances. To compensate for the potential inflation of Type I errors, the False Discovery Rate (FDR)—using the Benjamini–Hochberg method (Q = 5%)—was also determined. Comparisons with an FDR-adjusted p-value (q) less than 0.05 were considered discoveries.
4. Discussion
We have established an image analysis workflow to serve as a standardised method for the assessment of organoid viability and cytoarchitecture. The workflow was tested on a dataset of more than 1.5 million cells and demonstrated remarkable efficacy in identifying specified cell types and states, as well as providing temporal and spatial information when applied to cortical organoid development.
To enhance our analysis beyond single-plane imaging, we captured the entire organoid slice in 3D, revealing details of the cellular organisation that traditional maximum intensity projection methods omit. Additionally, our automated analysis of the complete section enabled us to explore the regional diversity within the organoids across each group. This comprehensive approach provided deeper insights into cellular features such as density, viability, and antibody expression at various developmental stages.
To implement high-throughput imaging, we utilised a widefield fluorescence microscope system. The advantage of this imaging approach was that it provided 3D acquisition of entire sections in a fraction of the time needed for confocal microscopy when using similar acquisition parameters. The widefield system adequately captured the nuclear marker and the peri-nuclear environment, which were the primary focus of our analyses. These findings suggest that widefield microscopy is a suitable alternative when a balance between resolution and higher acquisition speed is required.
The Leica Thunder system has three computational clearing algorithms available. Here, we implemented Small Volume Computational Clearing (SVCC). This algorithm applies adaptive deconvolution before removing unwanted background [
43]. Image datasets captured using this method provided sufficient cellular detail to facilitate automated cell counts. The resultant analysis workflow was designed to process widefield 3D image tiles; however, it could also be applied to any fluorescence image datasets for example, optically sectioned whole tissue samples using light-sheet microscopy [
19,
22]
At present, research using organoid models lacks a well-defined benchmark for ‘normal’ growth and development. We have established an “in-house” method to benchmark the elected organoid protocol tailored to the specific needs of cytoarchitectural analysis, an exercise critical to identify, interpret and compare deviations between control and disease models.
The analysis workflow’s initial task was to quantify non-viable and viable cells using the nuclear label. Non-viable or necrotic cells have previously been identified in organoid image-based studies by their higher fluorescence intensity and smaller diameter nuclei in comparison to functional cells [
50,
51,
52]. While previous studies focused on a single attribute, we adopted a more comprehensive approach by implementing a two-tiered Gaussian Mixture Model that analyses both volume and intensity. This approach allowed for more complex stratification of candidate cells. We show that nuclei classified using the integrated approach as non-viable cells was confirmed using cleaved-caspase 3 and TUNEL staining, consistent with previous reports [
12,
35,
46,
53] The approach also allowed automatic delineation of the non-viable core boundary from the viable region, which was a significant advancement.
To evaluate organoid viability, the nucleic viability classifications were applied to obtain quantitative “in-house” assessment of cortical organoids including the non-viable core volume and percentage non-viable cells in the viable region. We found a significant decrease in the volume of the non-viable core between 4 and 6 months post-induction. In contrast to this finding, previous research has proposed that the “dead cell” core increases over time, caused by the limited diffusion of oxygen and nutrients from the organoid surface in the avascular model [
14,
47,
54]. To interrogate the current finding, we quantified two additional factors likely to affect core size including the densities of viable and dead cells within the viable region. Our results indicated a reduced viable cell density within the viable region, from 4 to 6 months. Second, we found the viable region showed a decreased number of non-viable or dead cells from 4 to 6 months, which is consistent with reported cell death rates in cerebral organoids plateauing as they near synaptic maturity [
55]. Taken together, these results indicate diffusion rates across the viable region may be higher at 6 months. Additional transcriptional analysis would help to determine whether changes in gene expression involved in aerobic respiration and apoptotic genes are consistent with current findings [
56]. Overall, the results suggest that evaluating the non-viable core and viable region’s growth at a cellular level can help to understand how these factors might influence the health and development of cortical organoids in control and disease models.
To quantify the number of protein labelled cells, the analysis workflow was designed to capture each cell’s peri-nuclear protein expression level. Micro-structure in mature cortical organoids is complex in terms of both tightly packed heterogeneous cells and sub-cellular protein expression patterns, which make automating and obtaining cell counts for non-nucleic markers challenging. Therefore, our approach was to use each viable cell’s DAPI-mask and create a dilated 3D volume or peri-nuclear shell.
With respect to quantifying organoid maturation, our assessment of between 4 and 6 months post-induction showed no significant differences in S100β expression, indicating that astrocytic maturation remains stable throughout this period. This finding was somewhat unexpected, given the literature describing a continual increase in proliferation from the beginning of astrogenesis in cerebral models [
12,
21,
57,
58] Two possible explanations for these findings arise: first, it may be that the rate of astrocytic maturation is variable over time and that this variation has not previously been captured; second, although widely applied as a measure of astrocytic maturation, S100β is not expressed in all mature astrocytes [
40,
59] Therefore, quantitative changes in maturation based on S100β may be limited, and additional markers like glial fibrillary acidic protein (GFAP) could provide further insight.
The percentage of all cells characterised as mature MAP2 positive neurons was similar from 4 to 6 months post-induction, while the percentage of GABAergic neurons was significantly increased. These findings suggest a potential shift in the neuronal population towards GABAergic neuron s, consistent with previous studies that revealed a shift occurs at approximately 3–6 months post-induction [
9,
12] These observations emphasise how the image analysis workflow can be used to evaluate cell number in models of neurodevelopmental disorders, particularly where GABAergic interneurons are thought to have a significant impact [
9,
60,
61].
Future application of the image analysis workflow could include machine learning development. By capturing spatial and single-cell-level information from sample images, including protein signatures, the workflow can also be expanded to incorporate the cells’ molecular profile. This spatial mapping enables detailed profiling of each cell across large replicate datasets. Such advancements align with proposed developments in machine learning, where immunohistochemistry analysis of patient-derived organoids could serve as a valuable tool for personalised drug screening platforms [
62,
63]
In conclusion, we established a method to benchmark the preferred organoid protocol “in-house”. Functionalities were built into a single, customizable script and incorporated changes to improve their theoretical robustness, tailored to the specific needs of cytoarchitectural cortical organoid analysis. Implementing the current workflow would help laboratories to establish their own benchmark; a critical exercise for identifying, interpreting and comparing deviations across disease and control models. Furthermore, we hope that implementing this workflow in other laboratories using similar differentiation protocols will generate quantifiable outcomes, ultimately establishing a benchmark based on the approach itself rather than being limited to a specific lab.