
1. Introduction
The Bps of thousands of natural and synthetic analytes are unknown. Their Bp values are not only difficult to determine, but their determination would also be extremely expensive. Consequently, the boiling points, and most likely many other physicochemical parameters of analytes, seem not to be useful physicochemical measures to quantify isotope effects of analytes. On the other hand, data available for many other solvents, notably for purely inorganic water (Bp, 99.98 °C for H2O, Bp, 101.4 °C for D2O), suggest that the utility of the Bp would be of limited value.
δ(H/D) = tR(H) − tR(D)
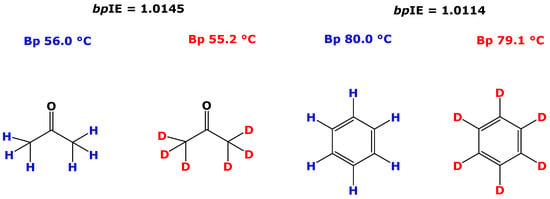
The underlying mechanisms of chromatographic isotope effects have been little explored. Many factors are likely to contribute to hdIEC, notably including differences in the physical and chemical properties of the isotopologs (e.g., C-H/C-D bond energies, boiling points), as described above for acetone and benzene. The lower Bp values of CD3COCD3 and C6D6 compared to those of CH3COCH3 and C6H6 could be, in analogy, due to somewhat weaker inter-molecular interactions of deuterated analytes compared to their protiated congeners.
ctEC = tR(cis)/tR(trans)
Again, based on the theory of the partition chromatography, the smaller retention times of trans-analytes compared to their cis-analytes would result from a weaker interaction strength of the trans-analytes with the GC stationary compared with the cis-analytes.
2. Methods
2.1. GC-MS Analyses Performed in the Author’s Group
2.2. GC-MS Analyses Performed by Other Groups
The experimental conditions of the work performed by other authors are reported in the Results section. For better understanding, the nomenclature used in the original articles was retained. The original data and the newly calculated hdIEC and ctEC values are presented in the Results section of the present work in form of Tables and Figures as appropriate.
2.3. Calculations and Data Presentation
hdIEC and c/tEC values were calculated using Formulas (2) and (4), respectively. GraphPad Prism Version 7 for Windows (GraphPad Software, San Diego, CA, USA) was used for the statistical analyses and preparation of graphs. Chemical structures of the investigated analytes and their derivatives were drawn using ChemDraw 15.0 Professional (PerkinElmer Informatics, Germany).
3. Results
3.1. GC-MS Analysis of Miscellaneous Analytes—Author’s Group
3.2. hdIEC in the GC-MS/MS Analysis of Fatty Acid Methyl Esters—Tintrop et al. 2022, 2023 [5,6]
The simultaneous analysis of FAMEs was performed on a GC 2010 with a MS/MS TQ8040 (Shimadzu Deutschland GmbH, Duisburg, Germany) in the MRM (SRM) mode. A Zebron ZB-FAME capillary column (30 m × 0.25 mm × 0.20 µm, Phenomenex, Torrance, USA) was used. The oven temperature program started at 40 °C, which was kept for 5 min, and then raised with a rate of 5 °C/min to 210 °C, where it was held for 5 min. Helium was used as a carrier gas (1.8 mL/min) and argon as a collision gas. Injection of the analytes was performed by splitless thermal desorption.
The H/D isotope effect is evident. Expectedly, the strongest chromatographic isotope effects are seen for the methyl esters of the perdeuterated fatty acids C16:0-d31 and C17:0-d33. The IEC values decrease with the increasing length of the fatty acid molecule. These observations suggest that the entire H/D effect of the d3Me group decreases with the increasing length of the fatty acid molecules by a factor of 10 (from 0.6% to 0.06% with respect to the retention time). According to the manufacturer’s information, Zebron™ ZB-FAME is based on a high-cyanopropyl (G48) chemistry (O-Si-(C3H6CN)n) and has high polarity. Analogous to the isotope effect seen in the GC-MS analysis of the methyl esters of amino derivatives, one may assume that the deuterium atoms of the d3Me ester group of the fatty acids weaken the interaction of the lipophilic fatty acid molecules with the immobilized liquid phase of the GC column. This effect is maximum in C16:0-d31 (IEC, 1.0171) and C17:0-d33 (hdIEC, 1.0126). The ctEC values for C18:1c and C18:1t are calculated to be each 1.0041 for d0Me and d3Me, i.e., they are higher than the hdIEC values of 1.0011.
3.3. hdIEC in the GC-MS Analysis of Fatty Acid-Pentafluorobenzyl Esters—Quehenberger et al. 2011 [4]
These observations suggest that the H/D isotope effect is evident in the GC-MS analysis of the FA-PFB ester derivatives. However, the isotope effect is weak. A possible explanation could be that the interaction of the PFB ester residue of the fatty acid derivatives with the stationary phase outweighs the interaction of the H/D atoms of the fatty acid skeleton.
3.4. Isotope Effects in the GC-MS Analysis of Cis- and Trans-Fatty Acid Pentafluorobenzyl Esters—Kuiper et al. 2018 [3]
Kuiper and colleagues developed a GC-NICI-MS method for the quantitative analysis of trans-fatty acids in human plasma, serum, and red blood cells (RBCs). As the trans-fatty acids coexist in biological samples at considerably lower concentrations, a “special” GC column known to allow simultaneous analysis of trans– and cis-fatty acids was used, i.e., an Agilent Select FAME.
Samples (100 µL) were combined with 100 μL of an internal standard (IS) solution that contained isotopologs. Subsequently, the samples were hydrolyzed (2 mL of 10% v/v 6 M HCl in acetonitrile followed by 2 mL of 10% v/v 10 M NaOH in methanol), each carried out at 104 °C for 45 min. After neutralization with 6 M HCl, the free FAs were extracted with hexane (three times, 2 mL each). The solvent was removed under vacuum (Genevac, Stone Ridge, NY, USA) and the samples were derivatized at room temperature for 15 min with 100 μL of 7% PFB-Br in acetonitrile and 10 μL triethylamine as the base catalyst. The fatty acid PFB (FA-PFB) esters were extracted with hexane (500 μL) and transferred to autosampler vials for GC-MS analysis. Samples were handled in glass vials to minimize contamination of samples with FAs from plastic supplies.
There are nine pairs of FAs and their 2H-labeled isotopologs. The FA-PFB derivatives of the unlabeled fatty acids had longer retention times than their 2H-isotopologs. The calculated hdIEC values ranged between 1.0428 for myristic acid (C14:0 and D27-C14:0) and 1.0009 for arachidonic acid (C20:4n-6,9,12,15 and D8-C20:4n-6,9,12,15).
These observations suggest that the FA-PFB ester derivatives of trans-fatty acids behave towards their cis-fatty acids in the same manner as behave non-deuterated towards deuterated fatty acids with respect to gas chromatography in fused-silica capillary columns. Note the long retention time range of the investigated FA-PFB ester derivatives in the study (i.e., 56 min to 116 min).
4. Discussion
Chromatographic separation of derivatized analytes in GC-MS is based on continuously occurring interactions of the analyte derivatives between a mostly silicone-based stationary phase (of small inner diameter and film-thickness) immobilized inside the GC column and mostly helium as the mobile phase. The just now “free“ analyte derivatives are “carried” by helium through the GC column until they are released into the ion-source of the GC-MS apparatus. The physicochemical properties of the analytes (e.g., boiling point, molecular weight, chemical structure, and possibly changing shape and orientation in the gas phase) contribute to the chromatographic effects, which are finally manifested in the retention times of the analytes. The retention time of analytes is a useful measure to quantify chromatographic effects. In quantitative GC-MS analyses, stable-isotope labeled analytes are used as internal standards.
Kinetic isotope effects, chromatographic isotope effects, and chromatographic cis/trans-effects have been known for several decades, but the underlying mechanisms are incompletely understood. Chromatographic isotope effects (IECs) are assumed to originate from differences in physicochemical properties of unlabeled and stable-isotope labeled analytes that result from the differences introduced into the analytes mainly by the heavy isotopes of H (hdIEC), C, N and O, i.e., 2H (D), 13C and 15N, respectively. Perhaps easier to understand are the chromatographic cis/trans-effects (ctEC), as the orientation of the molecules in the space may differ considerably, for instance in dependence on the residues in olefinic analytes R1-C=C-R2.
The retention time (tR) in gas chromatography (GC) and liquid chromatography (LC) is an experimentally ascertainable integral parameter, which incorporates all factors that are involved in the chromatographic process. In GC, they include boiling points vapor pressure, chemical composition, and three-dimensional structure of the analyte derivatives, the chemical composition of the stationary phase, adsorption/desorption processes, temperature of the injector port, initial and subsequently increasing temperature of the GC column and its dimensions (length, inner diameter, film thickness), as well as the nature and flow rate of the carrier gas. The present work addressed the issue of hdIEC and ctEC effects, and investigated these phenomena on a quantitative basis by re-examining data reported in the literature mainly by four groups including the author’s group. We hypothesized that the chromatographic isotope- and cis/trans-effects can be quantified by using the retention times of analyte derivatives in GC and introduced the parameters hdIEC and ctEC. The hdIEC and ctEC values were calculated by Formulas (2) and (4), respectively. The main working hypothesis was that both effects can be explained by differences in the interaction of the analytes with the stationary phase of fused-silica capillary GC columns. One class of analytes were amino acids that were analyzed as methyl ester pentafluoropropionic (PFP) derivatives. The second class free fatty acids that were analyzed as methyl ester (Me) or pentafluorobenzyl ester (PFB) derivatives.
The utilized GC columns differed in the chemistry of the stationary phase, and in part in length (15 m, 30 m, 200 m), had a comparable internal diameter, and small differences in film thickness(0.20 µm and 0.25 µm) of the immobilized stationary phase. The volume of the GC lumen was calculated to be about 0.7 mL, 1.5 mL and 9.6 mL, respectively. Under consideration of the reported carrier gas, and presumably laminar than turbulent flow, it is calculated that the carrier gas would need about 0.7 min to 5 min for a passage through the GC columns. The shape of the analytes inside the GC columns is unknown. In the case of hydrophobic analyte derivatives such as the FA-PFB derivatives, oblongness could possibly prevail. The interaction of the analyte derivatives and the stationary phase is considered to be due to different forces including van der Waals forces. Other effects, such as the organic solvent extract used in GC-MS analyses, “condensation” of analyte derivatives at lower GC column temperatures, and competition between the derivatives of analytes and their isotopologs, are possible but were not considered in this work.
4.1. Chromatographic Isotope Effects
The IEC values observed for PFB-O*NO2 and PFB-*NO2 (an asterisk * indicates 14N/15N; difference of 7% in atom mass) were very close to 1.0000 and indicate lack of a measurable IEC effect and virtually the same interaction extent of the central 14N and 15N atoms of PFB-O*NO2 and PFB-*NO2 due to sterical hindrance through the neighboring C and O atoms.
The IEC values observed for the 13C isotopologs of the fatty acid derivatives are almost identical to those of the 12C isotopologs, while the simultaneously analyzed 2H isotopologs clearly caused IEC. The C atoms of analytes do not interact directly with the stationary phase of the GC column. Furthermore, the 13C atoms of the 13C-labeled analytes differ by only 8% in atom mass from the 12C atoms. These two factors are likely responsible for the missing isotope effect in the 13C-labeled fatty acid derivatives.
The two N-methyl groups of dimethylamine (DMA) and metformin (Metf) in their PFBz, respectively, PFP derivatives caused strong hdIEC effects due to the six H/D atoms of the methyl groups which are not hindered sterically in their interaction with the stationary phase of the GC column. The comparably stronger hdIEC effects are likely to result from the greater differences in the mass of the H and D atoms (100% increase in atom mass).
whereas Bp(H) and Bp(F) are the boiling points of the protiated and perfluorinated solvent, respectively.
4.2. Chromatographic cis/trans-Effects in Fatty Acid Derivatives
The shape of the FA-PFB derivatives could be elongated for saturated fatty acids but curvy for unsaturated cis-fatty acids. Curvy fatty acids may possibly collide more often with the GC stationary phase through their –C=C-double bonds, thus increasing the residence time in the lumen of the GC column.
4.3. Chromatographic Effects Due to Other Types of Isomerism
4.4. Sterical Effects on Chromatographic Isotope Effects
4.5. Possible Implications of Chromatographic Isotope Effects in Metabolomics
5. Conclusions
The greatest IEC effects in isotopologs are exerted by the deuterium atoms of the lipophilic analytes, because they interact directly with the mostly hydrophobic stationary phase of fused-silica capillary GC columns. This interaction is weaker than that of the H atoms of the analytes, thus resulting in a weaker retention and shorter retention times of 2H-isotopologs. Even a small number of D atoms causes clearly and highly reproducibly quantifiable hdEC effects. Even thoroughly 13C-labeled analytes do not exert measurable IEC effects, because their “inner” C atoms do not interact directly with the stationary phase of the GC column and their possibly indirect action is rather negligible. This is likely to be true for 15N- and 18O-isotopologs even for derivatives such as PFB-*NO2, PFB-O*NO2, and FA-C*O2-PFB.
Source link
Dimitrios Tsikas www.mdpi.com
